











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Cancer is one of the main causes of morbidity and mortality worldwide. According to the World Health Organization, 9.6 million cancer deaths were reported in 2018, making it an important public health concern. Lung, prostate, and liver cancer are among the most prevalent in men and liver cancer is the second most common cause of death from cancer. Breast, cervical and thyroid cancer are among the most prevalent in women. In 2018, over 500,000 and 2 million of cases of cervical and breast cancer, respectively, were reported (WHO, 2019).
Cancer treatment commonly includes chemotherapy and radiotherapy; however, the anticancer drugs available have low selectivity and cause severe adverse effects, such as nephrotoxicity, neurotoxicity, and myelosuppression (Matsuo Lin, Roman & Sood, 2010). Therefore, the design and development of compounds as new anticancer agents against the types of cancer with the highest incidence are of vital importance in the area of health. In this sense, heterocyclic derivatives, such as quinoxaline (Kamal Bolla, Srikanth & Srivastava, 2013; Rivera et al., 2017a; Rivera et al., 2017b), uracil (Lu., Li, Mohamed, Wang & Meng, 2019), pyrimidines (Kilic-Kurt Bakar-Ates, Karakas & Kütük, 2018;Abdelhaleem., Abdelhameid, Kassab & Kandeel, 2018), β-lactams (Olazarán- Santibáñez, Bandyopadhyay, Carranza-Rosales, Rivera & Balderas-Rentería., 2017a; Olazarán et al., 2017b), and phthalimide (Bailly et al., 2003; Li et al., 2011) are used to obtain new anticancer agents (Figure 1).
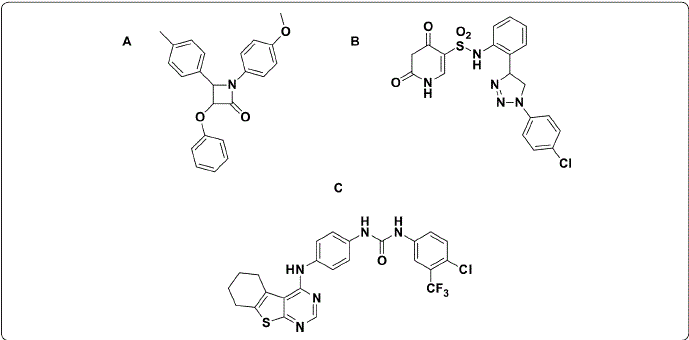
Figure 1 Chemical structures of compounds (A: β-lactam; B: uracil; and C: pyrimidin derivatives) with anticancer activity. Figure designed by the authors.
Phthalimide(Figure 2.A) derivatives arepromisingcompounds for the development of new anticancer agents (Li et al., 2011; Grigalius & Petrikaite, 2017; Kamal, Reddy, Reddy & Ramesh, 2002). However, the first step in knowing their potential as anticancer agents to determine their biological effects against cancer cell lines; for example, Bailly et al. obtained the phthalimide derivatives B and C (Figure 2.B and 2.C) with half-maximal inhibitory concentration (IC50) values of 0.6 μM and 4.9 nM, respectively, against CEM human leukemia cells (Bailly et al, 2003). Afterward, Al-Soud & Al-Masoudi (2001), reported compound D (Figure 2.D), a phthalimide derivative with 61% growth inhibition against the SF-268 glioblastoma cell line (Chen et al., 2010). Subsequently, Li et al. (2011) found that compound E (Figure 2.E) had higher IC50 values than the reference drug amonafide against the cancer cell lines MCF-7, HeLa, and 7721 (0.32, 1.02 and 0.46 µM versus 1.68, 1.73 and 4.27 µM, respectively).
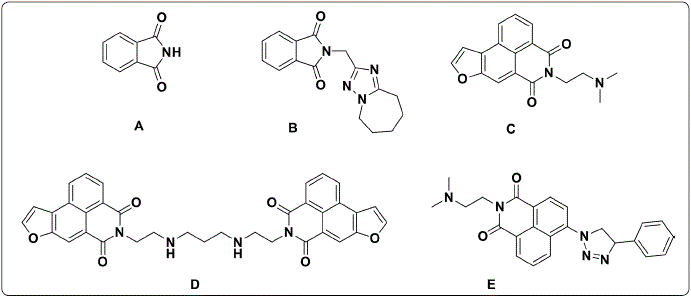
Figure 2 Chemical structure of phthalimide (A) and phthalimide analogs (B-E) with anticancer activity. Figure designed by the authors.
The mechanism of action of phthalimide derivatives as anticancer agents includes different drug targets, such as methyltransferases (Mai &Altucci, 2009;Asgatay et al., 2014), vascular endothelial growth factor receptor 2 (VEGR2) (Zahran et al., 2014; Othman, Gad-Elkareem, El-Naggar, Nossier & Amr, 2019; Philoppes & Lamie, 2019), lipoxygenases (LOX) (Aliabadi et al., 2015) fibroblast growth factor receptors (FGFRs) (Sundaresan et al., 2019) and topoisomerase II (Xie et al., 2011; Miyachi, Ogasawara, Azuma & Hashimoto, 1997; Yu & Wang, 2008). In the first decade of 2000, methyltransferases were reported as a promising target for cancer therapy (Siedlecki et al., 2006) and VEGR2 has been associated in in silico analyses with the biological effects of phthalimide derivatives. Based on the above, this work aimed to evaluate the antiproliferative activity of forty-three phthalimide derivatives against one main cancer cell line in men (HepG2) and two main cancer cell lines in women (HeLa, and 4T1). Additionally, the cytotoxicity of compounds against a normal murine fibroblast cell line (3T3) was determined. Finally, to determine their potential mechanism of action, a molecular docking analysis was done on the active site of two enzymes (DNMT1 and VEGR2) reported as the main potential phthalimide targets.
Materials and Methods
Chemistry
A total of forty-three phthalimide derivatives were obtained following the procedure reported by Kashif et al., (2018). The structural elucidation by infrared (IR) and proton nuclear magnetic resonance (1H-NMR) was done (Supplementary material).
Cell culture
The human cervical cancer (HeLa), human liver (HepG2), murine breast carcinoma (4T1) and murine fibroblasts (3T3) cell lines they were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were cultured in modified Dulbecco’s Eagle’s medium (DMEM/F-12) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antifungal solution (Sigma Aldrich, ST. Louis, MO).
Evaluation of proliferative activity by MTT
HeLa, HepG2, 4T1, and 3T3 cell lines were cultured at 5 x 103 cells/well, using flat-bottom 96 well plates and incubated overnight at 37 °C and 5% CO2.After, all phthalimide derivatives and doxorubicin (DOX) were added at 20 µg/mL(In Table I, the concentration for each compund were converted to micromolar units) previously diluted in DMEM/F-12 culture medium. Subsequently, the culture plates were incubated for 72 h. After this incubation, 20 µL of MTT/well (Sigma, St. Louis, MO, USA) was added to a final concentration of 5 mg/mLto quantify the percentage of proliferation. After 4 h of incubation, the supernatants were discarded and 100 µL dimethyl sulfoxide (DMSO) was added to each well to dissolve formazan crystals. Finally, measured at a wavelength of 540 nm using a Microplate Spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA). The percentage of cell proliferation was obtained through the following formula: Cell proliferation (%) = (Absorbance of treated cells/Absorbance of control cells) X 100.
Molecular docking analysis
A molecular docking analysis of phthalimide derivatives on the active site of human DNMT1 and VEGFR2 was performed using the software AutoDock Vina 1.1.2 (Trott & Olson, 2010). The crystallographic structures of DNMT1 in complex with S-adenosyl-l-homocysteine (SAH) and VEGFR2 in complex with methyl (5-{4-[({[2-fluoro-5-(trifluoromethyl)phenyl] amino}carbonyl)amino]phenoxy}-1h-benzimidazol-2-yl) carbamate (GIG) were retrieved from the Protein Data Bank (http://www.rcsb.org, PDB ID: 3PTA and 2OH4, respectively). The structure of both proteins was prepared using the Dock Prep tool of the software UCSF Chimera (Pettersen et al., 2004), which removes all ligands and water molecules of the receptor structure, also repairs missing side chains and adds polar hydrogens. Meanwhile, the co-crystallized ligands (SAH and GIG) and the phthalimide derivatives were prepared using Open Babel (O’Boyle et al., 2011); this tool minimized the structures of each ligand and adds polar hydrogens to them. Ligands and the receptor were converted to pdbqt format using MGLTools 1.1.6 (Morris et al., 2009). The docking search space had values of size X= 20, Y= 20, and Z= 20 and was centered according to the coordinates of the co-crystallized ligand in the PDB file. The non-covalent interactions of the docking results were calculated using Protein-Ligand Interaction Profiler (PLIP) (Salentin, Schreiber, Haupt, Adasme & Schroeder, 2015).
Results and Discussion
Antiproliferative activity
The results obtained from the evaluation of forty-three phthalimide derivatives against cell lines of the main types of cancer that cause mortality and normal cell lines to validate the selectivity of the biologicaleffect of thephthalimidederivatives at one concentration are shown in Table I and graphics 1-4. DOX was used a positive control. This drug is a chemotherapeutic agent with antiproliferative and cytotoxic effects, commonly used in various types of human cancer and biological models (Al-Abbasi et al., 2016).
Table I Biological effect (percentage cell proliferation) of phthalimide derivatives against cervical (HeLa), liver (HepG2) and breast (4T1) cancer and normal (3T3) cell lines at one concentration.

| |||||||
| Compound | R1 | R2 | Concentration (µM) | % Cell proliferation ± SD Panel/Cell Line | |||
| HeLa | HepG2 | 4T1 | 3T3 | ||||
| UTC | -- | -- | -- | 100.00 ± 3.00 | 100.00 ± 0.70 | 100.00 ± 1.01 | 100.00 ± 0.91 |
| H1 | H | C6H5 pOCH3 | 61.47 | 90.59 ± 1.32* | 99.18 ± 2.31 | 49.57 ± 2.09* | 62.33 ± 2.19* |
| H2 | H | C6H5 pCH3 | 64.65 | 96.18 ± 0.50 | 90.52 ± 0.52* | 98.13 ± 4.53 | 65.91 ± 1.48* |
| H3 | H | C6H5 pCH2CH3 | 61.85 | 92.88 ± 1.68 | 91.54 ± 0.97* | 84.64 ± 0.40* | 60.53 ± 1.27* |
| H4 | H | C6H5 pOH | 64.24 | 94.74 ± 4.60 | 97.04 ± 2.36 | 84.83 ± 0.40* | 67.26 ± 3.53* |
| H5 | H | C6H5 pNO2 | 58.77 | 89.95 ± 4.55* | 88.69 ± 2.00* | 86.20 ± 1.44* | 95.96 ± 3.39 |
| H6 | H | C6H5 pF | 63.84 | 98.73 ± 6.63 | 76.45 ± 2.64* | 97.61 ± 3.80 | 78.02 ± 3.53* |
| H7 | H | C6H5 pCl | 60.65 | 92.20 ± 2.84 | 84.61 ± 2.51* | 91.31 ± 8.94* | 43.94 ± 3.53* |
| H8 | H | C6H5 pBr | 53.44 | 99.66 ± 1.23 | 89.19 ± 0.85* | 98.55 ± 3.51 | 68.16 ± 0.70* |
| H9 | H | C6H3 oNO2O2C2H4 | 50.20 | 91.26 ± 1.59* | 100.0 ± 0.45 | 85.20 ± 1.75* | 69.05 ± 0.21* |
| H11 | H | C10H7 | 57.91 | 100.85 ± 3.49 | 90.11 ± 0.85* | 75.09 ± 0.86* | 48.87 ± 3.81* |
| H12 | H | C6H4 pC6H5 | 53.85 | 97.71 ± 3.30 | 84.51 ± 1.10* | 98.61 ± 4.61 | 24.21 ± 0.91* |
| H13 | H | C6H5 oNO2 | 58.77 | 86.77 ± 3.23* | 87.36 ± 1.16* | 75.52 ± 1.20* | 81.16 ± 0.77* |
| H14 | H | C4H3O | 70.11 | 88.55 ± 2.94* | 82.87 ± 0.88* | 85.97 ± 2.72* | 61.43 ± 0.21* |
| H16 | H | C4H3S | 66.37 | 91.26 ± 0.35 | 68.09 ± 2.36* | 94.01 ± 4.50 | 94.61 ± 0.21 |
| E1 | CH3 | C6H5 pOCH3 | 58.93 | 62.07 ± 1.10* | 99.24 ± 0.85 | 96.34 ± 4.53 | 47.98 ± 1.48* |
| E2 | CH3 | C6H5 pCH3 | 61.85 | 72.42 ± 1.15* | 90.52 ± 2.28* | 99.06 ± 2.20 | 95.51 ± 1.27 |
| E3 | CH3 | C6H5 pCH2CH3 | 59.28 | 77.23 ± 1.32* | 80.65 ± 1.00* | 98.89 ± 1.63 | 65.02 ± 0.70* |
| E4 | CH3 | C6H5 pOH | 61.47 | 70.50 ± 0.90* | 90.52 ± 2.75* | 97.61 ± 3.18 | 90.58 ± 1.13* |
| E5 | CH3 | C6H5 pNO2 | 56.44 | 94.59 ± 1.21 | 86.85 ± 3.26* | 82.52 ± 3.25* | 65.47 ± 0.49* |
| E6 | CH3 | C6H5pF | 61.10 | 85.76 ± 0.51* | 98.98 ± 1.51 | 98.80 ± 4.3 | 69.95 ± 1.42* |
| E7 | CH3 | C6H5 pCl | 58.18 | 98.25 ± 2.65 | 94.09 ± 1.36 | 80.43 ± 4.03* | 62.33 ± 3.46* |
| E8 | CH3 | C6H5 pBr | 51.51 | 96.73 ± 1.33 | 90.32 ± 4.23* | 100.64 ± 2.62 | 59.19 ± 0.07* |
| E9 | CH3 | C6H3 oNO2O2C2H4 | 48.50 | 81.26 ± 0.75* | 100.31 ± 1.67 | 93.38 ± 4.20 | 50.67 ± 0.35* |
| E11 | CH3 | C10H7 | 55.65 | 58.42 ± 1.65* | 100.0 ± 0.36 | 51.25 ± 0.30* | 78.47 ± 1.90* |
| E12 | CH3 | C6H4 pC6H5 | 51.89 | 60.02 ± 0.89* | 99.39 ± 1.32 | 91.11 ± 2.62* | 78.47 ± 1.69* |
| E13 | CH3 | C6H5 oNO2 | 56.44 | 66.26 ± 1.83* | 93.27 ± 2.60 | 95.77 ± 2.55 | 64.57 ± 1.34* |
| E14 | CH3 | C4H3O | 66.82 | 81.87 ± 0.40* | 92.56 ± 1.10 | 50.20 ± 1.04* | 77.57 ± 0.42* |
| E16 | CH3 | C4H3S | 63.42 | 58.57 ± 0.64* | 99.90 ± 2.51 | 51.23 ± 0.66* | 96.86 ± 4.17 |
| C1 | COOH | C6H5 pOCH3 | 54.15 | 83.78 ± 0.41* | 99.54 ± 0.15 | 98.26 ± 2.80 | 104.03 ± 0.70 |
| C2 | COOH | C6H5 pCH3 | 56.60 | 80.81 ± 0.15* | 97.96 ± 1.66 | 75.04 ± 1.24* | 69.05 ± 3.25* |
| C3 | COOH | C6H5 pCH2CH3 | 54.44 | 79.44 ± 0.55* | 100.20 ± 1.42 | 53.59 ± 2.65* | 79.82 ± 1.55* |
| C4 | COOH | C6H5 pOH | 56.29 | 73.04 ± 0.30* | 96.94 ± 3.53 | 50.26 ± 0.62* | 68.60 ± 0.49* |
| C5 | COOH | C6H5 pNO2 | 52.04 | 96.96 ± 1.85 | 94.90 ± 3.75 | 98.18 ± 3.15 | 68.16 ± 1.69* |
| C6 | COOH | C6H5 pF | 53.51 | 86.21 ± 2.82* | 100.0 ± 0.60 | 73.02 ± 2.10* | 59.19 ± 0.56* |
| C7 | COOH | C6H5 pCl | 55.97 | 63.67 ± 1.85* | 89.60 ± 3.21* | 58.18 ± 1.49* | 60.08 ± 2.54* |
| C8 | COOH | C6H5 pBr | 47.82 | 59.79 ± 0.73* | 100.15 ± 3.25 | 67.96 ± 2.86* | 87.44 ± 1.48* |
| C9 | COOH | C6H3 oNO2O2C2H4 | 45.21 | 79.51 ± 0.26* | 97.55 ± 2.88 | 64.84 ± 2.81* | 80.71 ± 1.13* |
| C10 | COOH | C6H3O2C2H4 | 51.21 | 82.79 ± 1.15* | 98.93 ± 0.55 | 100.54 ± 4.16 | 83.85 ± 0.63* |
| C11 | COOH | C10H7 | 48.14 | 78.37 ± 1.47* | 86.95 ± 1.61* | 92.87 ± 2.06 | 71.30 ± 1.34* |
| C12 | COOH | C6H5 pC6H5 | 52.04 | 86.14 ± 0.79* | 94.60 ± 1.88 | 92.87 ± 4.58 | 71.74 ± 0.14* |
| C13 | COOH | C6H5 oNO2 | 52.04 | 82.56 ± 2.28* | 99.80 ± 3.48 | 55.53 ± 0.37* | 60.98 ± 1.69* |
| C14 | COOH | C4H3O | 60.74 | 87.28 ± 0.43* | 83.49 ± 0.55* | 50.14 ± 0.77* | 60.53 ± 0.98* |
| C16 | COOH | C4H3S | 57.91 | 59.63 ± 1.01* | 94.29 ± 2.69 | 50.19 ± 0.62* | 72.64 ± 1.76* |
| DOX | - | - | 36.79 | 15.94 ± 2.60* | 50.37 ± 1.53* | 55.14 ± 0.96* | 40.97 ± 1.88* |
aData represent the means of triplicate samples with ± SD indicated. *p < 0.05 as compared with untreated cells (UTC).
Graphic 1 show that compounds from series H have a low effect on decreasing HeLa proliferation (0-15%), although, compounds H11 and H5, H9, and H13 show naphthyl and nitro groups that have been previously relationated with biological effects in anticancer agents, while the positive control DOX decreased cell proliferation by 84.06%. In general, the compounds from the second series E, with a methyl group on the phthalimide ring, significantly decreased HeLa cell proliferation, except for compounds E5, E7, and E8. Compounds E1, E11, E12, and E16 decreased HeLa cell proliferation by around 40%. These results indicate that the incorporation of a methyl group at 5-position on the phthalimide ring enhances their biological effect. Replacement of methyl by a carboxylic group in compounds of series C had a similar biological behavior on HeLa cells. Compounds C7, C8, and C16 significantly decreased the proliferation of the HeLa cell line (36.33, 40.21, and 40.37%, respectively). Interestingly, the compounds E16 and C8 not showed cytotoxic effects (proliferation percentage of around 96 and 87%, respectively) against the normal cell line (3T3) (Graphic 4) suggesting selectivity against the HeLa cell line. This antiproliferative activity from phthalimide derivatives on HeLa cell lines has been reported by Shiheido et al., (2012) They found that phthalimide derivatives such as 2-(2,6-diisopropyl phenyl)-5-amino-1H-isoindole-1,3- dione (TC11) inhibited cell proliferation of multiple myeloma lines (KMM1, KMM11, KMS27, KMS34, and RPMI8226) and induced caspase-mediated apoptosis on KMS34 and HeLa cell lines at a concentration of 50 µM.
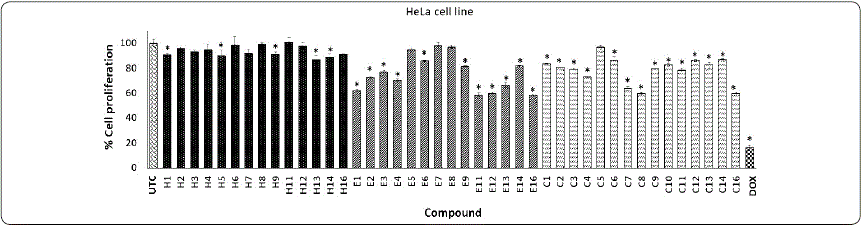
Graphic 1 Effect of phthalimide derivatives against HeLa cell line. Cervical cancer cells were treated with phthalimide derivatives and incubated for 72 h at 37 °C. Thereafter, an MTT assay was performed. The optical density was measured at 540 nm. Data represent the means of triplicate samples with ± SD indicated. *p < 0.05 as compared with untreated cells. Figure prepared by the authors.
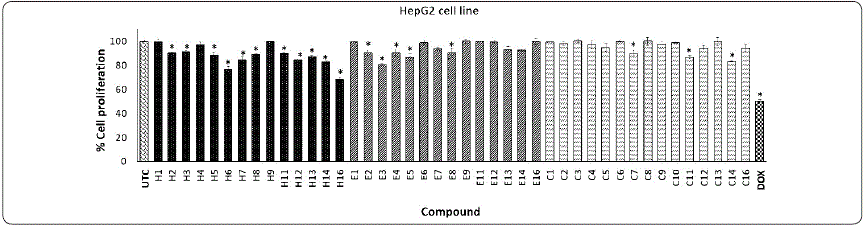
Graphic 2 Effect of phthalimide derivatives against HepG2 cell line. Hepatocarcinoma cells were treated with phthalimide derivatives and incubated for 72 h at 37 °C. Thereafter, an MTT assay was performed. The optical density was measured at 540 nm. Data represent the means of triplicate samples with ± SD indicated. *p < 0.05 as compared with untreated cells. Figure prepared by the authors.
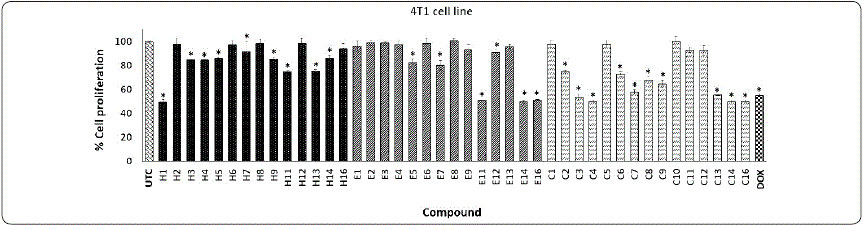
Graphic 3 Effect of phthalimide derivatives against 4T1 cell line. Breast cancer cells were treated with phthalimide derivatives and incubated for 72 h at 37 °C. Thereafter, an MTT assay was performed. The optical density was measured at 540 nm. Data represent the means of triplicate samples with ± SD indicated. *p < 0.05 as compared with untreated cells. Figure prepared by the authors.
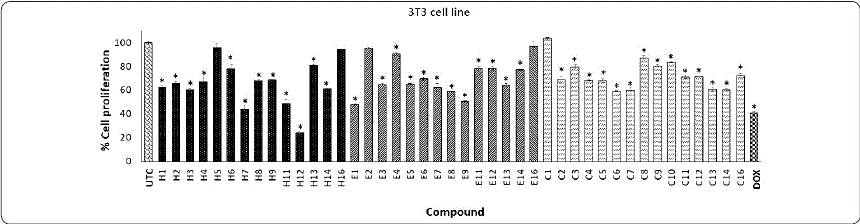
Graphic 4 Effect of phthalimide derivatives against 3T3 cell line. Murine fibroblast cells were treated with phthalimide derivatives and incubated for 72 h at 37 °C. Thereafter, an MTT assay was performed. The optical density was measured at 540 nm. Data represent the means of triplicate samples with ± SD indicated. *p < 0.05 as compared with untreated cells. Figure prepared by the authors.
Series H on HepG2 showed a low effect on the proliferative cell. Although, compound H16, with the thienyl group, decreased cell proliferation by 31.91% with a low effect (6%) on the 3T3 cell line. Subsequently, the incorporation of methyl or a carboxylic group on the phthalimide ring (series E and C, respectively) did not affect biological activity on the HepG2 cell line. In this same cell line, DOX decreased cell proliferation by 49.63% (Graphic 2). Similar to our results, Zahran et al., (2014) determined that the HepG2 cell line treated with phthalimide ester analogs after 48 h of incubation showed a moderate cytotoxic effect against the MCF-7 breast cancer cell line.
On the other hand, series H on 4T1 cells showed a null or low antiproliferative effect, except compound H1 (50.43%) (Graphic 3); however, these compounds also reduced the proliferative effect in normal cells (3T3) (Graphic 4). The incorporation of a methyl group on the phthalimide group reduced the antiproliferative effect (E1 with 99.24%). Graphic 3 shows that compounds E11, E14, and E16 on the 4T1 cell line demonstrated antiproliferative activity (47.75, 49.80, and 49.77%, respectively). Interestingly, E16 showed a low effect on normal 3T3 cells. These results indicate that E16 showed a selective effect by reducing proliferation in HeLa and 4T1 cells without significantly affecting the proliferation of 3T3 cells (Graphics 1, 3, and 4). Finally, series C against the 4T1 cell line showed the best results. Compounds C3, C4, C14, and C16 produced a good antiproliferative effect (from 45 to 50%). However, these compounds also affected the 3T3 cell line. DOX causes a decrease of 44.86% in cell proliferation against 4T1, and 59.03% against the 3T3 cell line. These results were similar to those obtained by Zahran et al., (2014) in which a macrophage cell line (RAW 267.7) was treated with 12.5 µg/mLof bis-phthalimide during 48 h of incubation, finding a cytotoxic effect of around 40%.
In summary, the results show that the HepG2 cancer cell line was the most resistant and that 4T1 was the most sensitive cancer cell line to the phthalimide derivatives evaluated. On the other hand, HeLa was more sensitive and 4T1 more resistant to DOX treatment (15.94% and 40.97% of cell proliferation, respectively).
Molecular docking analysis on DNA methyltransferase 1 To know the potential mechanism of action of phthalimide derivatives, a molecular docking analysis on the active site of DNMT1 with SAH andphthalimidederivatives was done. First, the non-covalent interactions in the crystallographic structure of the DNMT1-SAH complex (PDB ID: 3PTA) were calculated with protein-ligand interaction profiler (PLIP). As Figure 3 shows, nine hydrogen bonds were identified. These interactions show that the pyrimidine ring of SAH plays an important role in the binding to DNMT1 interacting with Met-1169,Asp-1190, and Cys-1191 through both of its nitrogens. In addition, the amine group attached to this ring interacts with the carboxyl group of Asp-1190.Another interesting subset of interactions are formed by Ssr-1146, Gly-1150, and Leu-1151 with an oxygen atom of the carboxyl group of SAH. The rest of the hydrogen bonds are formed with Asn-1578 and Ala-1579. This complex was also analyzed by docking to reproduce the crystallographic pose of SAH. The DNMT1-SAH-docked complex with the lowest vina score (-8.0 kcal/mol) had an RMSD of 1.603; an RMSD lower than 2 is generally considered adequate in silico reproduction.
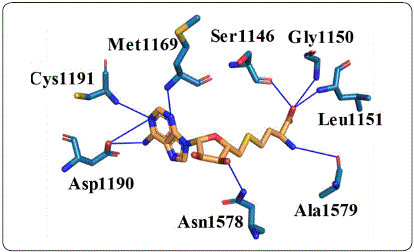
Figure 3 Non-covalent interactions between SAH and DNMT1. Hydrogen bonds are shown as blue lines. Figure designed by the authors.
The binding modes of the phthalimide derivatives were evaluated by molecular docking using vina. The poses with the lowest free energy of binding of each compound were selected and ranked. All the phthalimide derivatives showed an equal or greater binding affinity than SAH (≥ −8.0 kcal/mol). Table II shows the top ten compounds and highlights the structures that have lower free energy of binding. Biphenyl and naphthalene groups, as well as the benzodioxine group, were among the best- ranked compounds. These groups tend to generate hydrophobic interactions and stacking with the aromatic residues, Phe-1145 and Trp-1170, amino acids on the binding site of DNMT1. Four compounds from series C are in the top ten, maybe due to their extra carboxyl group, which can form hydrogen bonds easily.
Table II The top ten (free energy binding) of phthalimide derivatives on active site of DNMT1.
| Compound | Structure | Vina Score (kcal/mol) | Compound | Structure | Vina Score (kcal/mol) | |
|---|---|---|---|---|---|---|
| SAH |

|
-8.0 | C8 |

|
-10.1 | |
| C12 |

|
-10.5 | H12 |

|
-10.1 | |
| E12 |

|
-10.5 | H11 |

|
-9.7 | |
| C11 |

|
-10.4 | H8 |

|
-9.6 | |
| E8 |

|
-10.3 | C10 |

|
-9.5 | |
| E11 |

|
-10.3 |
To understand the affinity of these phthalimide derivatives with DNMT1, non-covalent interactions were calculated with PLIP. The compound with the lowest free energy of binding was C-12 (-10.5 kcal/mol), which forms hydrogen bonds with Gly-1150, Leu-1151, and Asn-1578, the same as SAH. There were also interactions with Glu-698, Cys-1148, Gly-1149, and Val-1580 via hydrogen bond. In addition to these interactions, the biphenyl group of C-12 has hydrophobic interactions with Phe-1145, Met-1168, Pro-1225, and Leu-1247. In addition, one of the rings of this biphenyl group forms a π-stacking with Phe-1145. This π-stacking interaction is also present in E12, which also has a free energy of binding of −10.5 kcal/mol, even with the loss of hydrogen bonds due to the lack of a carboxyl group. C12 and E12 also shared hydrophobic interactions with Met-1169 and Pro-1225 and the hydrogen bond withASN-1578. Interestingly, H12, which lacks the methyl group of E12, has a free energy of binding of −10.0 kcal/mol and four of its five interactions are the same in C12 and E12. These interactions are highlighted in Figure 4.
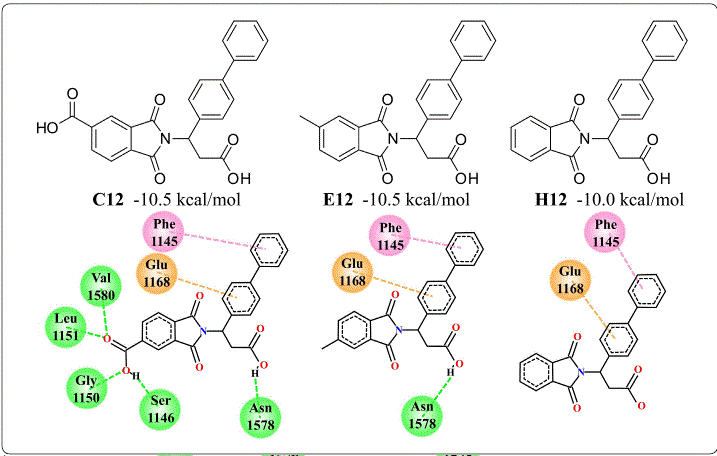
Figure 4 Structure and interactions of the compounds C12, E12 and H12 with a biphenyl group on the active site of DNMT1. This substructure showed to be the main source of energetic contribution to the affinity of the phthaloyl derivatives with DNMT1. Interactions are shown in green for hydrogen bonds, pink for π-stacking and orange for cation-π. Figure designed by the authors.
Another substructure of interest is the naphthalene group of C11 (−10.4 kcal/mol), E11 (−10.3 kcal/mol), and H11 (−9.7 kcal/mol), which keeps the π-stacking with Phe-1145 and hydrophobic interaction with Pro-1125 like the biphenyl group. These observations indicate that π-stacking interaction provides the greatest energy contribution. Another important contribution is by hydrogen bonds, in the comparison of interactions of H12 (−10.0 kcal/mol) and H11A (−9.7 kcal/mol), the only difference is a hydrogen bond with Asn-1578 of H12. Interestingly, benzodioxane does not generate π-stacking with Phe-1145 like biphenyl and naphthalene, nevertheless, the nitro group forms a hydrogen bond with Gly-1223 and Glu-1266, an interaction not present in other complexes. In conclusion, in silico calculations show that phthalimide derivatives interact with a higher affinity than SAH, a potent DNMT1 inhibitor. Non-covalent interactions of docked complexes point to a different inhibition mechanism between SAH and the phthalimide derivatives. In addition, it was noted that carboxyl and nitro groups contribute to binding through hydrogen bond formation, as well as biphenyl and naphthalene groups with π-stacking with Phe-1145. However, the antiproliferative activity from phthalimide derivatives is not relationed with binding on the active site of DNMT1; therefore, these results could suggest two options: phthalamide derivatives have low membrane permeability or they have another potential drug target.
Molecular docking analysis on VEGFR2
Previously, VEGFR2 a kinase receptor involved in angiogenesis has been propose as a drug target from phthalimide derivatives with anticancer activity using molecular docking (Othman et al., 2019). Therefore, in this study VEGFR2 was considering as a potential drug target. The 2HO4 pdb structure shows VEGFR2 in complex with GIG. The non-covalent interactions in this complex were calculated using the PLIP tool. Most of these interactions are hydrophobic and involved the residues Val846, Ala864, Lys866, Ile886, Ile890, Val897, Val914, Leu1033, Asp1044, and Phe1045. This complex also shows hydrogen bonding with Glu883, Cys917, and Gly920. To compare this complex with the possible binding mechanism of phthalimide derivatives, the molecular docking approach was applied first to GIG, obtaining a score of −10.4 kcal/mol, and then to the phthalimide derivatives. Table III shows the structure and score of the top ten compounds based on their vina score.
Table III The top ten (free energy binding) of phthalimide derivaties on active site of VEGFR2
| Compound | Structure | Vina Score (kcal/mol) | Compound | Structure | Vina Score (kcal/mol) | |
|---|---|---|---|---|---|---|
| GIG |

|
-10.4 | E5 |

|
-8.6 | |
| C12 |

|
-9.7 | C11 |

|
-8.9 | |
| E11 |

|
-9.1 | E6 |

|
-8.7 | |
| H12 |

|
-91 | E13 |

|
-8.6 | |
| E12 |

|
-8.9 | E8 |

|
-8.6 | |
| C5 |

|
-8.5 |
The molecular docking showed that none of the phthalimide derivatives had a better score than GIG. Nevertheless, these compounds shared some interactions with the complex VEGFR2-GIG. The carboxyl group, common in all the derivatives, is responsible for the hydrogen bonds with Cys917. In addition, C12, E12, and H12 have a biphenyl group that interacts hydrophobically with the same residues as GIG. C11 and E11 with their naphthalene ring interact in the same way as the biphenyl. Another similarity with GIG is the presence of halogen in the structure in the case of E8 and E6. Finally, C5 and E5, which share a nitro group, showed a hydrogen bond with Gly920, in addition to hydrogen bonding with Cys921. Although these results showed that some functional groups could interact with the residues on the binding site of VEGFR2, these compounds showed a lower docking score and a smaller number of non-covalent interactions than GIG. This suggests that these compounds may not target this protein efficiently.
Conclusions
Forty-three phthalimide derivatives against human cervical cancer (HeLa), human hepatoma (HepG2), murine breast carcinoma (4T1), and murine fibroblast (3T3) cell lines were evaluated. Compounds C16, E11, and E16 were the most antiproliferative agents against HeLa and 4T1 cell lines. In particular, compound H16 had a higher antiproliferative effect against the HepG2 cell line and a null effect against a normal 3T3 cell line. All phthalimide derivatives showed a lower free energy binding than SAH on the active site of DNMT1. However, the compounds showed higher free energy binding than GIG on the active site of VEGFR2. These results, with no correlation between the in silico analysis and the biological activity found in phthalimide derivatives, suggest two options: a) another potential drug target is involved in the mechanism of action, and b) poor physicochemical characteristics of compounds do not allow to cross cell membrane and hit the drug target. Therefore, more studies are needed to determine the biological effects that these kinds of compounds have on specific cell lines.

