











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Cystic fibrosis (CF), a monogenic disease transmitted in an autosomal recessive pattern, is multisystemic and chronic and originates as a consequence of pathogenic changes in the CFTR gene located on the long arm of chromosome 7 (locus 7q.31) that encodes for the protein known as the CF transmembrane conductance regulator (CFTR). Dysfunction of this protein causes altered ion transport in the apical membrane of epithelial cells in different organs. This dysfunction becomes evident in the early stages of life (infant stage). It is a complex and highly pleomorphic disease whose classic phenotype is a progressive obstructive pulmonary disease, exocrine pancreatic insufficiency, and elevated sweat chloride and sodium levels in 90% of patients1.
Epidemiology
CF is the most common inherited genetic disease among Caucasians; however, any ethnic group can be affected. Prevalence ranges from 1:1400 in Ireland to 1:3500 in the United States, while rates are much lower in geographic regions such as Asia and Africa2,3. In Latin America, the prevalence is 1:1600-14,000 live newborns (LNB), and in Mexico, 1:8500 LNB, with a detection rate of pathogenic variants of 41.6%4,5.
Mortality in childhood is due to complications of recurrent respiratory infections and malnutrition due to pancreatic insufficiency6. However, in Mexico and Latin America, life expectancy in the early 1990s averaged only 9 years. With the use of new therapies and better control of the disease, the average survival is 18 years1. In developed countries, life expectancy exceeds 40 years; these “long survivors” have a mean survival of 52.1 years of age in Canada, 47.4 years in the United States, and 47.3 years in the United Kingdom1,7.
Etiopathogenesis
CF is caused by pathogenic variants in the CFTR gene (locus 7q31.2) and is inherited in an autosomal recessive (AR) manner8-10. More than 2000 variants have been identified in the CFTR gene, but the most common variant is the phenylalanine deletion at position 508 (p.F508del, ΔF508)11,12. The pathogenic variants and their effects have been grouped into seven classes (Table 1)12-14.
Table 1 Classification of CFTR gene mutations. The most common mutations in each class and the type of defect are described
| Classification of CFTR mutations | Description | Examples | |
|---|---|---|---|
| Normal |  |
The CFTR protein is found on the cell surface and functions properly, allowing chloride and water transfer | — |
| Class I |  |
No protein production: No CFTR protein synthesis |
p.G542X p.W1282X p.R553X |
| Class II |  |
Traffic defects: CFTR protein is created but is misfolded, preventing it from reaching the cell surface |
p.F508del p.N1303K p.I507del |
| Class III |  |
Defective regulation: CFTR protein is created and reaches the cell surface, but there is a defect in the channel opening |
p.G551D p.S549N p.V520F |
| Class IV |  |
Decreased conductance: There is a defect in ion transport through the CFTR protein, resulting in a poor outflow of ions from the cell |
p.R117H p.D1152H p.R347P |
| Class V |  |
Reduced amount: CFTR protein is created but in insufficient quantities, resulting in decreased ion transport |
c. 621+1G>T c. 3849+10C>T c. 2780+5G>A p.A455E c. 3140-26A>G |
| Class VI |  |
Increased turnover: A decrease in the half-life of the CFTR protein occurs, causing accelerated turnover of the CFTR protein |
c. 4326delTC p.Gln1412X |
| Class VII |  |
No mRNA: No mRNA is synthesized, no CFTR protein is formed |
Dele2,3 (21kb) |
CFTR, cystic fibrosis transmembrane conductance regulator; mRNA, messenger ribonucleic acid.
Structurally, CFTR is a membrane-bound glycoprotein with a typical 12-helix architecture comprising two pseudosymmetric transmembrane domains (MSD) and two nuclear binding domains (NBD) that bind and hydrolyze ATP. Between both NBD units is a single regulatory domain (R) consisting of many charged amino acids (Figure 1)15. The CFTR protein is expressed mainly in the lung and the pancreas, sweat glands, intestine, liver, nasal mucosa, salivary glands, and reproductive tract16. Under normal conditions, CFTR works like a gate tightly coupled to ATPase cycles (Figure 1) through NBD dimerization, which subsequently induces the formation of a transmembrane domain cavity that opens towards the extracellular side to allow the flow of selective anions as chlorine (Cl-) and bicarbonate (HCO3-)11.
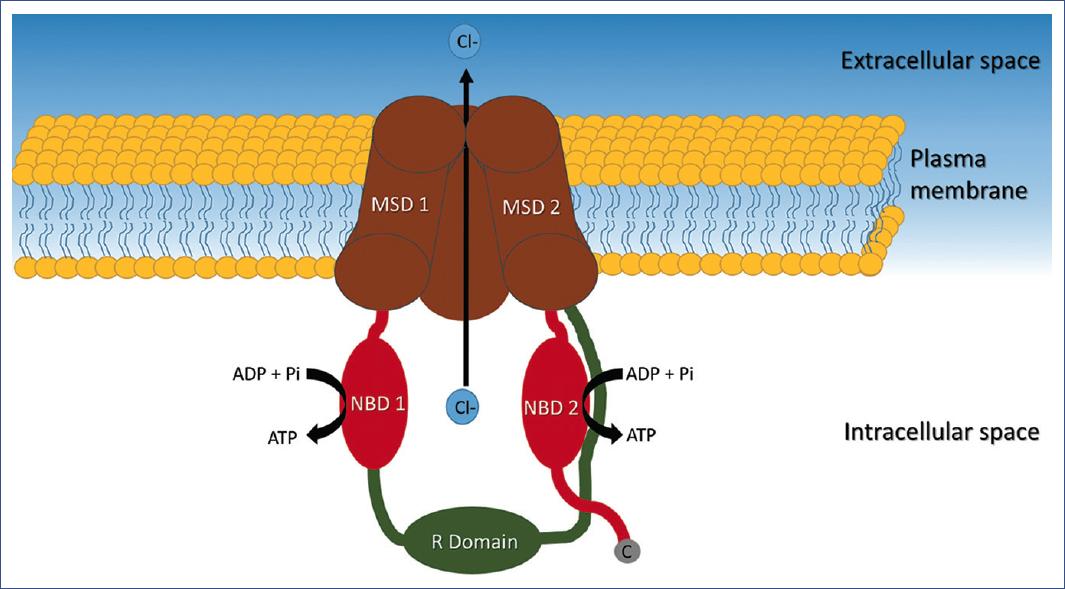
Figure 1 Representation of the CFTR protein channel, composed of two transmembrane domains (MSD 1 and 2), each bound to a nucleotide-binding domain (NBD 1 and 2). NBD1 is connected to MSD2 by a regulatory domain (R). NBD 2 has a carboxyl end. CFTR, CF transmembrane conductance regulator; MSD, transmembrane domain; NBD, nucleotide-binding domain; R, regulatory domain.
Particularly in the airway, CFTR regulates the local pH by allowing Cl- and HCO3- to flow out of the cell; in addition, it causes the epithelial sodium channel ENaC to transport sodium into the cell (Figure 2). Normal airway surface fluid (ASF) has a pH around 7.017. However, in CF, the pH is eight times more acidic due to the lack of adequate secretion of Cl- and HCO3- anions into the extracellular space (Figure 2). The loss of chloride secretion due to CFTR deficiency results in changes in osmotic pressures and electroneutrality, which probably lead to excessive sodium and water absorption18.
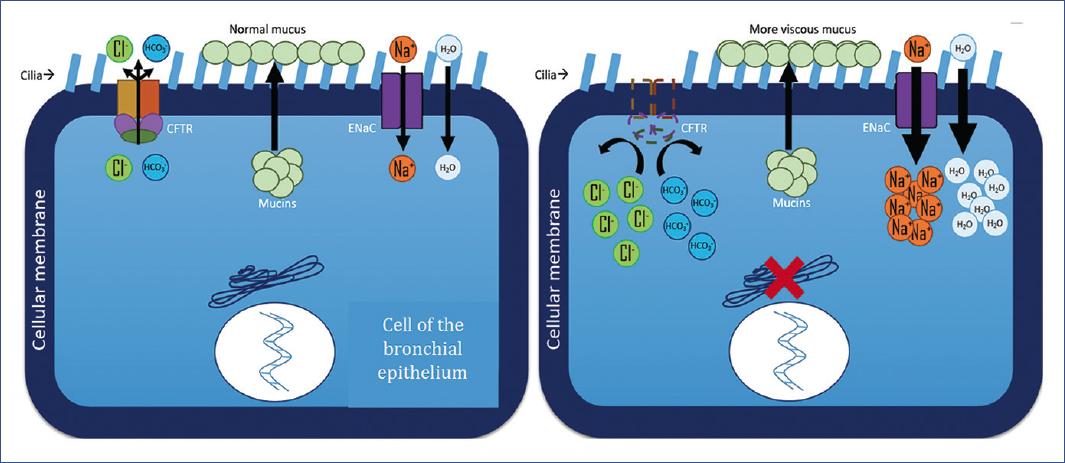
Figure 2 The function of the CFTR channel, the function of the ENaC channel, and mucin secretion for the formation of normal airway surface fluid are illustrated. A. Cell dynamics in a healthy context. B. Cell dynamics in CF in the context of the p.F508del mutation. The intradomain defect affects full-length CFTR protein assembly and post-translational stability, whereby deletion of phenylalanine at position 508 in NBD1 leads to a CFTR trafficking defect. CFTR, cystic fibrosis transmembrane conductance regulator; ENaC, epithelial sodium channel.
Pulmonary alterations are due to the addition of a considerably more acidic pH (caused by the lack of secretion of Cl- and HCO3- anions) and the excessive absorption of Na+ and water that cause an increase in mucus viscosity and a difficult clearance of mucus in the airway. In the other organs and systems, CF produces obstructions at different levels, causing extrapulmonary symptoms.
Clinical manifestations
Respiratory, gastrointestinal, and metabolic alterations are present in CF, some associated with inadequate nutrient uptake related to intestinal pathology.
The presenting symptoms may vary according to age at diagnosis and are sometimes related to genotype. Prior to the implementation of newborn screening in the United States, the diagnosis of CF in infants and preschoolers was integrated after presenting with one or more of the following symptoms19: respiratory symptoms (45% of patients), inadequate weight for age (28%), or meconium ileus (20%). In addition, the clinical presentation in adults with atypical features is better in the long term than those diagnosed in childhood19.
Signs and symptoms can be grouped according to the age group or location of the damage. According to age group, meconium ileus, prolonged neonatal jaundice (cholestatic), edema, anemia, malnutrition, steatorrhea, malabsorption syndrome, inadequate weight gain, and recurrent vomiting may be found in newborns and young infants. There is often recurrent or chronic coughing or wheezing in infants that do not improve with treatment, recurrent or chronic pneumonia, growth retardation, chronic diarrhea, rectal prolapse, salty skin taste, and chronic hyponatremia, and hypochloremia. Preschool children may show chronic cough with purulent expectoration and chronic recurrent unexplained wheezing (both with no response to treatment), poor weight and height gain, recurrent abdominal pain, rectal prolapse, intussusception, chronic diarrhea, clubbing of the fingers, chronic hyponatremia, and chronic hypochloremia, hepatomegaly or unexplained liver disease, and nasal polyps. Schoolchildren present with chronic unexplained respiratory symptoms, Pseudomonas aeruginosa in bronchial discharge, chronic sinusitis, nasal polyposis, bronchiectasis, chronic diarrhea, distal intestinal obstruction syndrome, pancreatitis, rectal prolapse, and hepatomegaly. Moreover, adolescents and adults often present with chronic unexplained suppurative lung disease, digital clubbing, recurrent abdominal pain, pancreatitis, distal intestinal obstruction syndrome, liver cirrhosis and portal hypertension, growth retardation, male sterility with azoospermia, and decreased fertility in women20. These symptoms are characteristic in patients with chloride concentrations > 60 mmol/L (discussed later). However, a subgroup of patients with chloride concentrations in the intermediate range is diagnostically challenging. As mentioned by Jorquera21, these patients may develop minimal symptoms or other symptoms related to defects in the CFTR gene in non-respiratory organs, making diagnosis difficult. Symptoms usually range from clubbing toes, recurrent respiratory infections, and isolation of P. aeruginosa and Staphylococcus aureus. Less common symptoms are pancreatic insufficiency, meconium ileus, distal intestinal obstruction syndrome, and liver disease21.
Respiratory symptoms
Respiratory alterations can be found in 80% of infants and preschoolers19. In children under one year of age, dry and repetitive cough is frequent. Also, persistent tachypnea, a slight increase in the anteroposterior diameter of the thorax, decreased expansion of the upper thorax, persistent intercostal retraction, and bronchial obstruction may be observed. Older children may present with bronchial obstruction and cough with purulent mucous secretions as constant symptoms. There are variable degrees of thoracic deformity with increased anteroposterior diameter. In the thoracic auscultation, in general, there is no added noise in intercrisis periods, and crackles in infectious exacerbations are present in children with severe compromise. In advanced disease, bronchiectasis, with or without hemoptysis, watch glass nails, and clubbing fingers may be present, while cyanosis is a late sign20.
Sinus disease is also present in most patients and manifests as chronic nasal congestion, headache, cough, chronic posterior nasal discharge, and sleep disorders19.
With increased viscosity and difficult clearance, respiratory secretions obstruct the airways and promote infection, leading to tissue destruction and, eventually, bronchiectasis. In chronic airway obstruction, Haemophilus influenzae, S. aureus, P. aeruginosa, and Burkholderia cepacia are the main pathogenic bacteria22.
Some germs have been implicated in the infection that occurs in CF. Before the antimicrobial era, few children with CF survived beyond the age of 2 years. Children died primarily from failure to thrive, along with bacterial pneumonia. The organism found in the lungs at autopsy in these children was almost always S. aureus, which remains the predominant pathogen of CF from birth to adolescence, with 80% of children infected in preadolescence and early adolescence. Moreover, the critical pathogen in chronic lung disease is an unusual morphotype of P. aeruginosa termed mucoid. Although this germ is comparatively avirulent, it induces a chronic inflammatory response, apparently responsible for the lung damage that leads to the patient's demise. This process may occur over years or decades, which would explain why 60-75% of adults in the United States are infected with this organism. In other cases of CF, a double count of glucose non-fermenting bacilli that are B. cepacia complex organisms, Achromobacter and Stenotrophomonas, are more frequently found in the patient. The latter is found in approximately 10% to 20% of CF patients, while Achromobacter is found in 5% to 10%. The incidence of these organisms increases with age and probably reflects patients who have received multiple rounds of antipseudomonal antimicrobial therapy23.
In the case of P. aeruginosa, responsible for morbidity and mortality in CF, a prevalence of 25-30% has been reported in infants aged 0-1 year, 38-40% in children aged 2-6 years, and can reach 80% in patients > 18 years. Infection is mainly caused by rough strains with high sensitivity to antibiotics. However, in the case of CF, this infection is not completed and acquires a mucoid phenotype in response to alteration of the CFTR gene epithelium. Therefore, the stagnation of mucus due to dehydration helps form an ideal culture medium and a layer around the bacteria that makes the immune system and antibiotics less effective. Consequently, endobronchial infection becomes chronic and can no longer be eliminated24.
The most frequently detected symptoms in the more advanced stages are pulmonary hypertension, bronchiectasis, and cor pulmonale. Progressive lung disease remains the leading cause of morbidity and mortality in these patients22.
Signs and symptoms that evidence pulmonary exacerbation are increased cough, changes in sputum (consistency or volume), decreased appetite, or weight loss. Exacerbation also shows changes in respiratory examination and respiratory rate. However, the number and variability of signs and symptoms may alter the exacerbation pattern25.
Extra-respiratory symptoms
Digestive system and nutrition. About 3.9% of patients do not present extrapulmonary manifestations related to pancreatic, gastrointestinal, and hepatobiliary dysfunction26,27. Defects in the CFTR gene result in abnormalities affecting organs such as the liver, gastrointestinal tract, and pancreas. Nutritional failure in CF is multifactorial. Malabsorption of lipids, protein, and fat-soluble vitamins results from insufficient pancreatic enzyme production, which bile salt abnormalities may aggravate in the presence of concurrent liver disease. Because of the predisposition to pulmonary infection, a relationship has been found with increased breathing, reduced appetite, and increased calories at the expense of inflammatory catabolism. Approximately 85% of patients develop pancreatic insufficiency between 1-2 years of age. In the context of CF, pancreatic insufficiency is a risk factor for recurrent pancreatitis, which is the presentation for the diagnosis of CF26.
Gastrointestinal tract manifestations are related to mucosal dysmotility and include meconium ileus, constipation, distal intestinal obstruction syndrome, gastroesophageal reflux disease, and small intestinal bacterial overgrowth. The hepatobiliary disease has a prevalence of 10-15%, with peak onset in preadolescence.
In exclusively breastfed infants, the triad of anemia, hypoalbuminemia, and edema—secondary to malnutrition—and some specific deficiencies due to malabsorption caused by CF are common20.
CF-related liver disease. Approximately 20-40% of people with CF develop clinically detectable CF-related liver disease (CFLD)19,26.
Hypovitaminosis. CFLD and pancreatic dysfunction lead to lipid malabsorption, predisposing to deficiencies of fat-soluble vitamins (A, D, E, and K)28.
CF-related diabetes (CFRD). The main cause is an insulin deficiency related to the destruction of pancreatic islets29. Two percent of CF patients have CFRD in childhood, increasing to 20% of adolescents and reaching 40-50% of CF patients in adulthood. This increasing proportion suggests multifactorial, complex, and progressive pathogenesis of CFRD. Excessive protein catabolism is problematic in CF lungs, associated with an imbalance between catabolic and anabolic enzymes.
Studies in adults have shown that insulin depletion contributes to an inflammatory catabolic state, compromising lung function30. It has been suggested that CFRD patients with no impaired basal glycemia do not require treatment because they are asymptomatic. However, the nutritional consequences of insulin deficiency may impair their quality of life. In the case of patients with altered basal glycemia, microalbuminuria was observed in 14%, retinopathy in 16%, neuropathy in 55%, and gastropathy in 50%. Therefore, from the age of 5 years, it is advisable to monitor annually for microvascular complications from the diagnosis of CFRD31.
Recurrent venous thrombosis. CF seems to be a risk factor for the development of recurrent venous thrombosis. Of 120 children and young adults with acute venous thromboembolism, 19 showed recurrent thrombosis. Among them, six had CF, of which five were infected with B. cepacia32.
Anemia. It occurs in 10% of children but is more frequent in older adults or those with decreased pulmonary function. Although its causes are diverse, the relationship with CF remains unclear, especially in infants, in whom it may occasionally be a presenting sign. Mechanisms involved include dysregulation of iron metabolism, anemia due to chronic disease, blood loss, renal failure, or bone marrow suppression after transplantation28.
Bone disease. Bone disease, characterized by decreased mineral density and increased rates of fracture and kyphosis, is rare in children under 10 years of age but increases in prevalence with age. In addition, vitamin D deficiency decreases intestinal absorption of calcium and produces secondary hyperparathyroidism, which leads to increased bone resorption and fragility1.
Infertility. Most male CF patients (> 98%) have a congenital bilateral absence of the vas deferens (CBAVD) and are infertile, but < 2% remain fertile and usually carry CFTR genotypes. It has been proposed that reduced CFTR activity impairs ion exchange within the male genital ducts and predisposes to increased intraluminal viscosity and obstruction by accumulated secretions and mucus, ultimately leading to obliteration of epididymis and vas deferens33,34. In contrast, women with CF experience more pulmonary exacerbations after puberty than men and increased colonization with the mucoid form of the commonly acquired CF pathogen P. aeruginosa. Gonadal hormones may contribute to these sex differences. Female sex hormones (estrogen and progesterone) are expressed in lung tissue and their hormone receptors. There is a 35% subfertility and infertility rate in women with CF trying to conceive (mean age 30.4 years). In the cervix, the defective CFTR in the epithelium promotes the production of thick, dehydrated cervical mucus. This mucus blocks the cervix and impairs the entry of spermatozoa. In the endometrium, defective CFTR in the epithelium can alter bicarbonate secretion, reducing uterine fluid volume and impairing sperm fertilization capacity. Finally, women with CF may have a lower ovarian reserve compared to age-matched controls35.
Nephrolithiasis and nephrocalcinosis. CF has been associated with nephrolithiasis and nephrocalcinosis due to factors that promote calcium deposition.
Diagnosis
New challenges have emerged in both the diagnosis and clinical management of children with CF, as various forms of terminology for the disease have been introduced in recent years. During the development of the Cystic Fibrosis Foundation guidelines, newborns diagnosed with CF were identified by screening through the analysis of sweat chloride levels with values ≥ 60 mmol/L36.
The primary test for confirming the diagnosis of CF is the sweat chloride test, performed according to the guidelines and standards recommended by the Cystic Fibrosis Foundation committee, which places this test as the current gold standard for diagnosing CF. Moreover, it is appropriate to maintain the cut-off values recommended by the committee based on the available data from chloride testing in healthy and CF-affected infants with the following ranges for infants up to 6 months of age: ≤ 29 mmol/L, unlikely CF; 30 to 59 mmol/L, intermediate; ≥ 60 mmol/L, indicative of CF37,38.
Currently, the extensive use of immunoreactive trypsin (IRT) neonatal screening has aided in diagnosing CF in asymptomatic or low symptomatic patients in approximately 64% of new diagnoses in the United States. Current guidelines on early diagnosis recommend sweat chloride testing by the Gibson and Cook technique in patients with positive screening37. There is a low probability of CF diagnosis if the result is < 30 mmol/L. However, in the case of an intermediate-range (30-59 mmol/L), the test should be repeated two to three weeks later, or consider extended CFTR gene analysis or CFTR functional analysis to classify the case as CFTR-related metabolic syndrome (CRMS)/CF-screening positive, inconclusive diagnosis (CFSPID). When in both occasions the result is > 60 mmol/L, the diagnosis of CF is confirmatory, always being careful to interpret false positives (caused by conditions such as malnutrition or Down syndrome, among others) or false negatives (due to technical errors, hypoproteinemia, or sampling of the newborn in the first days). Generally, these false results are due to technical errors. Further CF functional tests, such as nasal potential difference or intestinal absorption tests, can be performed. However, these should be performed in specialized centers certified by the CF foundation36.
Newborn screening for CF depends on the initial identification of elevated immunoreactive trypsinogen (IRT) values in the blood of the newborn. Since normal IRT reference values vary slightly, after identifying an abnormal IRT value, most programs perform DNA testing to detect known CFTR gene mutations (IRT/DNA strategy). This strategy provides a sensitivity of approximately 90-95% and identifies newborns at risk for a broad spectrum of disease severity36.
Once the screening result is positive, sweat chloride analysis should be performed in newborns > 36 weeks of gestation and > 2 kg of weight. Genetic testing should be performed as soon as possible when available38. The definitive diagnosis is based on the criteria listed in Table 233.
Table 2 Diagnostic criteria for cystic fibrosis
| Presence of clinical criteria (at least one): |
| Organ with consistent symptoms of cystic fibrosis |
| Chronic sino-pulmonary disease |
| Characteristic gastrointestinal and nutritional abnormalities |
| Salt loss syndromes |
| Obstructive azoospermia |
| Sibs with cystic fibrosis |
| Positive test result at birth (positive neonatal screen) |
| Tests showing CFTR dysfunction (at least one): |
| CFTR dysfunction indicated by elevated levels in sweat |
| chlorine analysis (> 60 mmol/L) in two tests |
| Nasal potential difference consistent with cystic fibrosis |
| Presence of two pathogenic variants in CFTR in different alleles |
CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator.
Adapted from Brennan et al.33
The diagnostic algorithm proposed in the guidelines of the CF Foundation (2017)38 (Figure 3) and the one proposed by the clinical practice guideline in Mexico5 (Figure 4) are published and available for review.
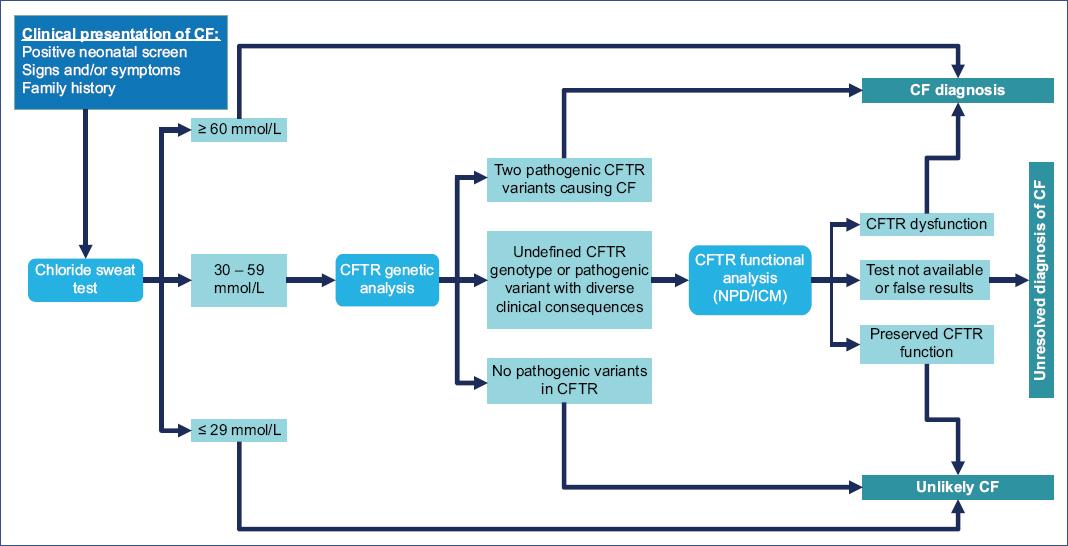
Figure 3 Diagnostic algorithm for individuals with high suspicion of cystic fibrosis. CFTR, CF transmembrane conductance regulator; NPD, nasal potential difference; ICM, intestinal current measurement.
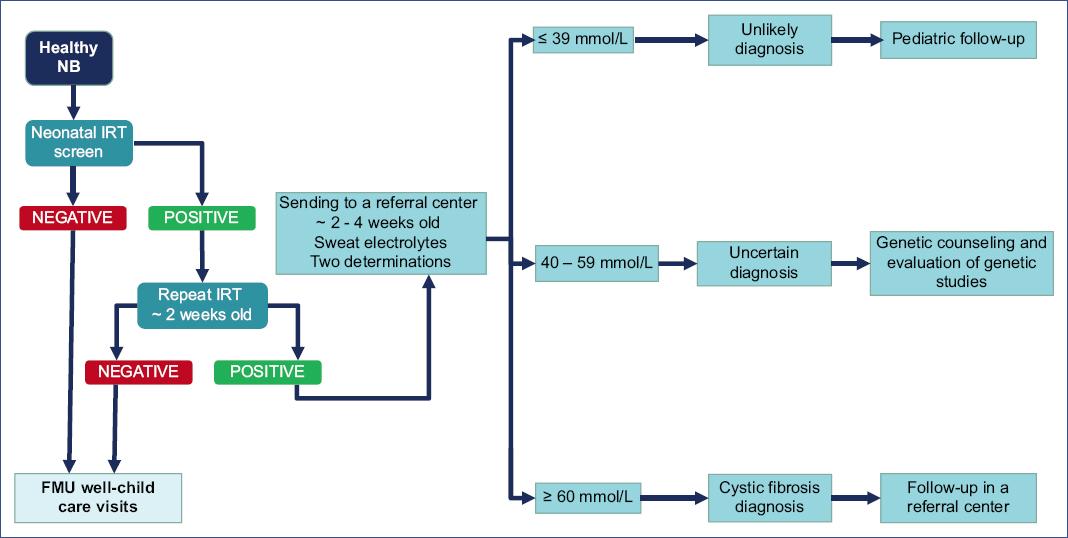
Figure 4 Cystic fibrosis diagnosis algorithm. NB, newborn; FMU, family medicine unit; IRT, immunoreactive trypsin.
Pulmonary function tests help assess disease severity and progression. Spirometry can be performed in children aged 3-6 years using modified acceptability criteria, and most children aged ≥ 6 years can perform reliable lung volume tests39. Most patients with cystic fibrosis develop an obstructive pattern. The first manifestations of early obstruction are an increase in the ratio of residual volume to total lung capacity ratio and a decrease in forced expiratory flow of 25-75% of lung volume. Later, in advanced disease, there is a decrease in forced expiratory volume in one second (FEV1) and FEV1/FVC (forced vital capacity) ratio40.
Chest X-rays and computed tomography are the imaging studies used in CF. Patients are usually monitored with chest X-rays every 2 years. Radiographic findings change according to the progression of the disease: hyperinflation, which later becomes persistent, can be observed. Bronchovascular markings are also visible and may progress to a pattern of bronchiectasis and cyst formation. Also, a progressive flattening of the diaphragms is present. The alterations usually appear first in the upper lobes.
CT is used when detailed knowledge of the extent of lung disease is required. High-resolution CT is useful in asymptomatic children who are small for pulmonary function testing; findings may include peribronchial thickening, air trapping, bronchiectasis, centrilobular nodules41. Pulmonary function changes may not be closely related to abnormalities found on CT (progressive).
When inconclusive results are not obtained with sweat chloride and DNA testing, the nasal potential difference can be measured to assess CFTR dysfunction33. The potential difference is measured by placing electrodes in the nasal cavity and comparing the voltages between baseline and administration of amiloride to block sodium transport and after nasal perfusion with a chloride-free solution, such as isoproterenol, to stimulate the CFTR-dependent chloride transporter. There is a potential difference in individuals with CFTR abnormalities at baseline, which decreases after amiloride and shows a minimal response to perfusion with chloride-free isoproterenol42.
Finally, a gut malabsorption study can be performed. The pancreatic exocrine function can be assessed indirectly by measuring fecal elastase. A low fecal elastase level suggests pancreatic insufficiency and consequent gut malabsorption43.
Management
The management of CF is multidisciplinary and involves both the patient and family members, who will facilitate follow-up and optimization of treatment. The following are the three pillars of treatment that should be considered a guideline for all CF patients.
Keep the airway clear of secretions
Maintaining a clear airway at all times can be achieved through various techniques and schemes that can be combined according to the specific characteristics of each patient. They can be divided into two main groups:
a. Physical therapy. It seeks to clean the airways and drain the accumulation of thick, viscous secretions that can harbor bacteria and their products. Respiratory physiotherapy is performed in two or more daily sessions depending on the severity and complications, separated by a minimum of 2 hours after meals44. It has been shown that the application of this therapy can have short-term benefits for patients with CF. However, there are as yet no conclusions on the long-term benefits45.
Techniques vary and depend on age: in children < 3 years of age, passive chest percussion maneuvers, blocks, vibrations, and postural drainage are used. After 3 years of age, forced expiration techniques can be added, inducing the child to exhale slowly and progressively, to bring secretions into the central airway. In children > 6 years, autogenic drainage maneuvers are taught37.
Active cycle of breathing techniques, autogenic drainage, and Vojta approach can be combined with respiratory reeducation exercises to improve diaphragmatic function by reducing the number of respiratory cycles but with a higher tidal volume. In addition, physical activities that stimulate respiratory training, such as swimming, cycling, or athletics, can be implemented. One of the advantages of using specialized techniques such as physiotherapy in poorly treated patients with or patients with little or no physical activity is that it has helped to find new independent and effective ways of managing respiratory secretions. Physical exercise of different types influences the quality of life, sensitivity, and resistance to lung infections46.
In addition to physical therapy, there are mechanical aids for the elimination of secretions. Among them, the following stand out:
a) Positive expiratory pressure (PEP) mask. This mask is resistant to expiration.
b) Oscillating positive expiratory pressure (Flutter) devices. These devices combine both PEP and oscillation, generating vibrations that separate bronchial secretions throughout the airway.
c) High-frequency chest compression. This percussion vest generates pressure on the thorax through a pump, but it is costly.
d) Percussive intrapulmonary ventilator. This ventilator includes the administration of a continuous aerosol together with internal percussion of the thorax through small bursts of air at 200-300 cycles per minute44.
b. Pharmacological therapy
a) DNase I (alpha dornase). This endonuclease breaks the DNA chains released by neutrophils responsible for the high viscosity of secretions in CF. It has been shown to improve lung function, increasing FEV1 by 6% in CF patients > 6 years and decreasing the frequency of pulmonary exacerbations. The usual dose is 2.5 mg to nebulize every 24 hours, daily and permanently, after using aerosol bronchodilators (albuterol) to prevent bronchoconstriction47.
b) Hypertonic saline solution at 7%. Pre-glucocorticoid nebulization with saline has been used to overcome hydration failure of the aqueous layer of the bronchial mucosa. This treatment reduces the frequency of exacerbations in symptomatic respiratory patients > 12 years. In children < 12 years of age, insufficient evidence has been found to justify its routine use since, in children > 6 years of age, no improvement in the frequency of exacerbations was observed. However, in tracheostomized pediatric patients, cleaning with 5% saline solution before initiating nebulizations reduces the occurrence of exacerbations37. The use of inhaled hypertonic saline is suggested in young children based on randomized trials in which lung clearance index is measured and compared to placebo (isotonic saline)48. In a randomized trial in infants < 4 months, Stahl et al. found that inhaled hypertonic saline twice daily for 52 weeks demonstrated a significant change in lung clearance index compared to isotonic saline49. Similar results were observed in another randomized trial in children aged 36-72 months50.
Keep the airway free from infections
Respiratory tract infections are persistent. The administration of antibiotics, together with physical therapy and pharmacological agents that allow the elimination of secretions, will facilitate the control of the CF patient. The following is a description of the antibiotics recommended for the four main etiologic agents in respiratory tract infections:
a) S. aureus is the predominant pathogen in the first years of life and the first to infect the respiratory tract. Treatment is based on oral dicloxacillin or flucloxacillin, or intravenous dicloxacillin in exacerbations.
b) P. aeruginosa causes the most severe chronic pulmonary infection and is associated with progressive deterioration of respiratory function. Treatment consists of oral ciprofloxacin or ceftazidime, intravenous aminoglycosides or antipseudomonal penicillin, and intravenous aminoglycoside in severe cases for 21 days.
c) B. cepacia is associated with more significant pulmonary deterioration in adolescents and adults. The incidence in Latin America is low, and treatment is based on temocillin with an aminoglycoside.
d) H. influenzae can cause chronic colonization. The indicated treatment is amoxicillin-clavulanic acid or second or third-generation cephalosporin51.
Currently, intravenous therapy has been replaced by inhaled treatments, which allow prolonged treatments with high doses and minimal toxicity. The recommended drugs are tobramycin, colistin, and aztreonam-lysine, which should subsequently be inhaled beta-blockers such as salbutamol to prevent bronchoconstriction37.
Maintain an optimal nutritional state
There is a close and bidirectional correlation between body mass index (BMI) and lung function (FEV1). The goal of nutritional therapy is to maintain a BMI above the 50th percentile37.
Evidence has shown that CFTR dysfunction impairs β-cells, leading to a decrease in insulin levels. Since insulin from β-cells can also exert an inhibitory effect on glucagon release from α-cells, this suggests a dual role of CFTR in regulating glucagon secretion and may contribute to glucose intolerance in CF, leading to some conditions. In the plasma of CF patients, elevated levels of peroxidized lipids and oxysterols are observed, indicating abnormal lipid metabolism and increased susceptibility to lipoprotein lipid oxidation. In addition, pancreatic insufficiency and decreased bile acids lead to malabsorption of critical lipid-soluble antioxidants, such as carotenoids, tocopherols, and coenzyme Q-1030.
The elements for adequate management of nutritional status in the patient with CF are the following: a) evaluation of BMI or the weight/height ratio in all patients; b) enzyme replacement therapy in pancreatic insufficiency; c) replacement therapy of lipid-soluble vitamins (A, D, E, K); d) hypercaloric and hyperproteic regimen to maintain metabolic goals; e) maintenance of bone health; f) replacement of bile salts in case of liver disease; g) treatment of diabetes mellitus.
Management in a specialized multidisciplinary center
It is recommended that CF patients be managed in a specialized center with the following characteristics and staff: CF specialists, a team of support specialists, a coordinating nurse, infrastructure, laboratories, and integrated outpatient care.
Children with CF regularly require hospitalization, either electively or due to acute pulmonary exacerbations or deterioration of their health status. Criteria for hospitalization are increased frequency of productive cough, increased amount or changes in the presence of sputum, increased respiratory rate, dyspnea at rest, hypoxia, marked decrease in a vesicular murmur, new changes in chest X-ray, deterioration of pulmonary function tests, weight loss or inadequate weight gain, hemoptysis52. As the patient progresses to advanced lung disease, options for care and prognosis should be discussed. Among these options is lung transplantation, which should be of both lungs to avoid foci of infection that could damage the transplanted lung.
The following are the referral thresholds recommended by the Cystic Fibrosis Foundation:
-
– In patients > 18 years:
FEV1 < 50% of the predicted value and rapidly declining
FEV1 < 40% of the predicted value and any of the following markers of shortened survival: > 2 exacerbations per year requiring intravenous antibiotics, massive hemoptysis > 240 mL requiring intensive care unit admission or bronchial artery embolization, pneumothorax, BMI < 18
FEV1 < 30% of the predicted value
-
– In patients < 18 years:
-
– In all patients, any of the following, regardless of FEV1:
6-minute walk test < 400 m
Hypoxemia (SpO2 < 88% or PaO2 < 55 mmHg) at rest or with exertion
Hypercarbia (PaCO2 > 50 mmHg) confirmed on arterial blood gases
Pulmonary artery systolic pressure > 50 mmHg on echocardiogram or evidence of right ventricular dysfunction in the absence of tricuspid regurgitant jet
Any exacerbation requiring positive pressure ventilation
-
– Other factors to be considered for the possibility of earlier transplantation, even when other thresholds are not met, are as follows:
Female sex, especially younger females
Short stature (height < 162 cm)
Liver cirrhosis or chronic kidney disease may require consideration of multiple-organ transplantation and may affect the timing or choice of transplant center53.
Current therapies
At present, therapies to correct changes in the CFTR gene are focused on preventing a specific pathogenic variant from restoring messenger RNA levels (class I variants), correcting the folding and trafficking of CFTR to the apical plasma membrane (correctors for class II variants), or increasing CFTR channel function (enhancement therapy for class III variants and any variants with residual function at the apical plasma membrane). Other therapies in preclinical development are directed toward non-specific variants and include the following: gene therapy with viral and non-viral vectors (nanocarriers such as liposomes, dendrimers, exosomes); genome editing using zinc finger nuclease (ZFN) systems, effector nucleases such as transcription activators (TALENs) or CRISPR/Cas9; antisense oligonucleotides or small interfering RNAs to selectively inhibit ENaC expression; or stem cell therapy to repair airway tissue. Based on these options, personalized and more effective therapy for the CF patient can be determined14,54,55.
Personalized treatment strategies include:
Agents that act through reading target premature stop codon changes, such as aminoglycosides, although they are limited due to their ototoxicity and nephrotoxicity. NB124 restored 7% of CFTR function in respiratory cell lines with G542X, R553X, R1162X, and W1282X variants; PTC124 (Atularen®), for the G442X variant, showed mixed results and no improvement, and RCT101 used nanoparticle-transported suppressor transfer RNA56.
Calcium-activated chloride channel agents (CaCCs) increase intracellular calcium release. These agents include duramycin and denufosol14.
ENaC inhibitors block sodium channels used in combination with modulators. First-generation inhibitors include amiloride; second-generation inhibitors include benzamil and phenamil; third-generation inhibitors include GS-9411 and SPX-101. Camostat also indirectly inhibits ENaC by reducing Na+ transport through pulmonary epithelial cells14.
-
CFTR modulators are drugs that correct protein folding, trafficking to the plasma membrane, or CFTR protein function caused by specific pathogenic variants in the CFTR gene. They are divided into the following groups:
– Enhancers. They increase the probability of channel opening and enhance CFTR function, such as ATP analogs, IBMX (3-isobutyl-1-methylxanthine), genistein, PT1-808, GLPG1837, QBW251, and VX-770. VX-770 or ivacaftor prolongs the duration of channel opening, improves ion transport through protein channels making secreted mucus more fluid, decreases respiratory symptoms and the number of severe respiratory infections, increases FEV1 and weight, and decreases sweat chloride. It is authorized to treat CF caused only by specific pathogenic variants44,54,57,58.
− Correctors. They improve the folding defect, cellular processing, and trafficking of CFTR through the endoplasmic reticulum to the apical plasma membrane and increase CFTR expression on the cell surface, such as the chaperones 4-phenylbutyrate, curcumin, miglustat, and FDL169. VX-809 or lumacaftor increases chloride secretion by 14-25% for p.F508del homozygotes, but without clear clinical improvement, so some studies combine it with VX-770 (Orkambi®), although without adequate efficacy. VX-661 (tezacaftor) is also used for the p.F508del variant in homo or heterozygous state with variants with residual function, and combined with VX-770 (Symdeko®), a mean absolute improvement of 6.8% was obtained. Other correctors are currently being investigated, such as elexacaftor either alone or in double or triple combination (Trikafta®), apparently with better results in CFTR function44,54,59.
– Amplifiers. These agents stimulate CFR protein expression and are applied in class V variants. They can be used in combination with enhancers and correctors such as PTI-42854.
– Stabilizers. These molecules rectify protein instability, increase CFTR time at the plasma membrane, and decrease its degradation in the endoplasmic reticulum, such as hepatocyte growth factor, S-nitrosoglutathione (GSNO), and cavosonstat (N91115, an inhibitor of S-nitrosoglutathione reductase for p.F508del homozygotes)54,60,61.
In conclusion, the measurement of IRT in the neonatal screen has allowed early diagnosis of patients with CF. However, these efforts imply challenges to homogenize diagnostic criteria and achieve more timely management, including the three pillars of treatment, to improve the quality and survival of CF patients. Research also offers new challenges and ethical issues due to its scope, but above all, a cure through precision medicine, a dream whose achievement we believe is not far off.

