











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
It is well known that boronic acids react with 1,2 and 1,3 diols to form 5- or 6-membered cyclic boronic esters, respectively, having the Boron atom in a trigonal planar geometry [1,2]. The boronic acids and diols interactions have received special interest because of their high stability and the directionality offered by the B-O covalent bonds, as well as the reversibility of the process in the presence of an external stimulus such as water molecules, acid or basic media, which promote the hydrolysis of the boronic esters [3-5]. In fact, the characteristics mentioned above, have been exploited in a great variety of applications such as sugar sensing [6-10] or host-guest chemistry [11-13], functional polymers [14], tissue histology [15], NMR shift reagents [16,17], molecular self-assembled materials [18,19], fluorescence imaging carbohydrates [20] and dynamic covalent bonding [21].
A prominent chemical feature of the trivalent boron compounds is the presence of an empty p-orbital which is responsible for the Lewis acidity of boron [19], providing the capacity to form coordinative bonds with a series of donor ligands [22,23]. For instance, trisubstituted boron compounds react with N-donor ligands forming N→B coordinative bonds, here the boron atom changes its coordination number to tetracoordinate producing a tetrahedral geometry. The N→B coordinative bond has resulted in a powerful tool for constructing supramolecular architectures such as macrocycles, capsules, and polymers [24-41]. Previously, we described the synthesis of imine boronic esters by multicomponent reactions and demonstrated their ability to sense metal cations conditioned by the presence of a pyridine fragment in the molecular structure [42-44]. In order to get more information about the formation of boronic esters combined with other functional groups such as carbonyl and amine moieties, herein, we report the synthesis and structural analysis of zwitterionic compounds containing boronates which were obtained by esterification reactions.
Experimental
Materials
All reagents and solvents were acquired from commercial suppliers and used without purification.
Instrumentation
The 1H, 13C and 11B NMR spectra were recorded at room temperature using a Varian Gemini 200 or Varian Unit 400 spectrophotometers. TMS (internal, 1H, δ = 0.00 ppm, 13C, δ = 0.0 ppm) and BF3 .OEt2 (external 11B, δ = 0.0 ppm) were used as standard references. Two-dimensional COSY and HETCOR experiments were carried out for unequivocal signals assignment. The NMR spectra were processed using the MestReNova software [45]. Infrared spectra were recorded on a Nicolet 6700 FT-IR Thermo Scientific spectrophotometer. Mass spectra were obtained with a Jeol JMS 700 equipment. Melting points were determined with a Büchi B-540 digital apparatus.
X-ray Crystallography
X-ray diffraction studies for compounds 1b, 1d, 1f and 2e were performed on a Bruker-APEX diffractometer with a CCD area detector, using Mo Kα-radiation, (λ = 0.71073) and a graphite monochromator. Frames were collected at T = 293 K, by ω/ϕ-rotation (Δ/ω = 0.3°) at 10 s per frame. The measured intensities were reduced to F 2. Structure solution, refinement and data output were carried out with the OLEX2 program package [46] using SHELXL-2014 [47] for refinement. Non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed in geometrically calculated positions using a riding model. Intermolecular distances were analyzed with Mercury [48].
General method for the preparation of heterocyclic boronate esters 1a-1h and 2e
Compounds 1a-1h were synthesized from the equimolecular reaction between the corresponding arylboronic acid derivatives and 2-amino-2-methyl-1,3-propanediol or serinol in methanol (methanol/acetone for 2e). For compounds 1a-1h, the reaction mixtures were stirred for 1 h at room temperature and let stand overnight. The formed white precipitate was collected by filtration, showing low solubility in most common organic solvents. The final products were purified by recrystallization or washing using methanol as solvent. For compound 2e the reaction was stirred under reflux for 3 h. The product was obtained as yellow crystals after slow evaporation of the solvent.
Compound 1a was synthesized from 0.05 g (0.33 mmol) of 4-formylphenylboronic acid and 0.035 g (0.33 mmol) of 2-amino-2-methyl-1,3-propanediol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.070 g (91%); m.p.(decomp) > 300 °C. IR (ATR) ν (cm-1): 3054 (O-H, m), 2864 (N-H, w), 1697 (C=O, m), 1597 (w), 1516 (m), 1381 (w), 1303 (w), 1209 (m), 1172 (m), 1098 (s), 1050 (s), 988 (s), 890 (s), 817 (s), 674 (m). EI-MS m/z (%): 219 ([M-H2O]+, 52), 161 (55), 147 (32), 97 (35), 57 (67). 1H NMR (200 MHz, D2O) δ: 9.82 (1H, s, H-C=O), 7.81 (2H, d, J = 7.7 Hz, H-3, H-5), 7.69 (2H, d, J = 7.7 Hz, H-2, H-6), 3.53 (4H, AB, J = 11.0 Hz, H-7, H-9), 1.14 (1H, s, H-10). 13C NMR (50 MHz, D2O) δ: 197.1 (C=O), 133.9 (C-4), 131.8 (C-2, C-6), 129.0 (C-3, C-5), 64.1 (C-7, C-9), 56.4 (C-8), 17.8 (CH3). 11B NMR (128.4 MHz, D2O) δ: 2.0 ppm (h 1/2 = 2133 Hz).
Compound 1b was synthesized from 0.05 g (0.33 mmol) of 3-formylphenylboronic acid and 0.035 g (0.33 mmol) of 2-amino-2-methyl-1,3-propanediol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.053 g (69%); m.p. = 195-200 °C. IR (ATR) ν (cm-1): 3092 (O-H, m), 2876 (N-H, w), 1696 (C=O, m), 1572 (w), 1523 (w), 1376 (w), 1311 (w), 1225 (w), 1105 (m), 1058 (s), 988 (s), 898 (s), 829 (s), 792 (s), 699 (s). EI-MS m/z (%): 219 ([M-H2O]+, 100), 187 (59), 147 (39), 114(12), 95(15), 69(23), 57(15). 1H NMR (200 MHz, DMSO-d 6) δ: 9.53 (1H, s, H-C=O), 8.07 (1H, s, H-2), 7.71 (1H, d, J = 7.6 Hz, H-4), 7.62 (1H, d, J = 7.4 Hz, H-6), 7.29 (1H, t, J = 7.6 Hz, H-5), 4.31 (4H, AB, J = 10.7 Hz, H-7, H-9), 1.07 (1H, s, H-10). 13C NMR (100 MHz, DMSO-d 6) δ: 190.5 (C=O), 136.3 (C-2), 135.7 (C-3), 133.3 (C-6), 130.9 (C-4), 128.3 (C-5), 68.7 (C-7, C-9), 59.5 (C-8), 21.1 (CH3). 11B NMR (128.4 MHz, D2O) δ: 5.0 ppm (h 1/2 = 960 Hz).
Compound 1c was synthesized from 0.05 g (0.304 mmol) of 4-acetylphenylboronic acid and 0.032 g (0.304 mmol) of 2-amino-2-methyl-1,3-propanediol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.068 g (89 %); m.p. = 113-119 °C. IR (ATR) ν (cm-1): 3086 (O-H, m), 2873 (N-H, w), 1667 (C=O, m), 1597 (w), 1524 (w), 1393 (w), 1360 (w), 1279 (w), 1205 (w), 1185 (w), 1107 (m), 1054 (s), 993 (m), 862 (s), 825 (s), 747 (m). EI-MS m/z (%): 233 ([M-H2O]+,100), 191 (25), 186 (29), 161 (53), 114 (14), 57 (17). 1H NMR (200 MHz, D2O) δ: 7.86 (2H, d, J = 7.9 Hz, H-3, H-5), 7.63 (2H, d, J = 7.9 Hz, H-2, H-6), 3.54 (4H, AB, J = 12 Hz, H-7, H-9), 2.61 (3H, s, CH3-), 1.15 (1H, s, H-10). 13C NMR (50 MHz, D2O) δ: 204.9 (C=O), 134.5 (C-4), 131.7 (C-2, C-6), 127.4 (C-3, C-5), 63.8 (C-7, C-9), 56.9 (C-8), 26.7 (CH 3 CO), 17.6 (CH3). 11B NMR (128.4 MHz, D2O) δ: 3.0 ppm (h 1/2 = 2560 Hz).
Compound 1d was synthesized from 0.05 g (0.304 mmol) of 3-acetylphenylboronic acid and 0.032 g (0.304 mmol) of 2-amino-2-methyl-1,3-propanediol in 10 mL of MeOH. The product was obtained as white powder. Yield 0.065 g (85%); m.p. = 75-79 °C. IR (ATR) ν (cm-1): 3091 (O-H, m), 2863 (N-H, w), 1666 (C=O, m), 1585 (w), 1503 (w), 1462 (w), 1430 (w), 1397 (w), 1356 (w), 1258 (m), 1238 (m), 1144 (d), 1111 (s), 1054 (s), 1017 (m), 984 (m), 878 (s), 796 (s), 764 (s), 697 (s). EI-MS m/z (%): 233 ([M-H2O]+, 100), 201 (30), 186 (24), 161 (40), 114 (14), 57 (15). 1H NMR (200 MHz, DMSO-d 6) δ: 8.00 (1H, s, H-2), 7.95 (1H, d, J = 7.0 Hz, H-4), 7.64 (1H, d, J = 7.0 Hz, H-6), 7.52 (1H, t, J = 7.6 Hz, H-5), 3.5 (4H, broad, overlapped with water signal, H-7, H-9), 2.56 (3H, s, CH3-), 2.42 (1H, s, H-10). 13C NMR (50 MHz, DMSO-d 6) δ: 198.7 (C=O), 136.5 (C-2), 135.6 (C-3), 133.3 (C-6), 131.4 (C-4), 128.8 (C-5), 71.5 (C-7, C-9), 66.7 (C-8), 26.7 (CH 3 CO), 26.5 (CH3). 11B NMR (128.4 MHz, DMSO-d 6) δ: 1.5 ppm (h 1/2 = 1882 Hz).
Compound 1e was synthesized from 0.05 g (0.33 mmol) of 4-formylphenylboronic acid and 0.03 g (0.33 mmol) of serinol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.0505 g (68 %); m.p.decomp = 344 °C. IR (ATR) ν (cm-1): 3105 (O-H, m), 2849 (N-H, w), 1671 (C=O, m), 1557 (w), 1476 (m), 1419 (m), 1312 (s), 1244 (s), 1160 (s), 1114 (m), 1064 (m), 1018 (m), 830 (m), 726 (m), 646 (s). EI-MS m/z (%): 205 ([M-H2O]+, 100), 173 (34), 147 (58), 100 (15), 77 (11). 1H NMR (200 MHz, D2O) δ: 9.85 (1H, s, H-C=O), 7.77 (4H, m, H-2,6, H-3,5), 3.58-3.86 (5H, m, H-7, H-8, H-9). 13C NMR (50 MHz, D2O) δ: 197.2 (C=O), 134.1 (C-4), 132.2 (C-2, C-6), 129.0 (C-3, C-5), 59.5 (C-7, C-9), 53.8 (C-8). 11B NMR (128.4 MHz, D2O) δ: 6.6 ppm (h 1/2 = 358.5 Hz).
Compound 1f was synthesized from 0.05 g (0.33 mmol) of 3-formylphenylboronic acid and 0.03 g (0.33 mmol) of serinol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.045 g (60 %); m.p. = 320-322 °C. IR (ATR) ν (cm-1): 3066 (O-H, m), 2861 (N-H, w), 1686 (C=O, m), 1596 (w), 1543 (w), 1371 (m), 1257 (m), 1199 (m), 1049 (s), 865 (s), 786 (s), 700 (s), 647 (s). EI-MS m/z (%): 205 ([M-H2O]+, 32), 175 (100), 119 (22), 91(22), 57(33). 1H NMR (200 MHz, DMSO-d 6) δ: 9.90 (1H, s, H-C=O), 8.11 (1H, s, H-2), 7.93 (1H, d, J = 7.5 Hz, H-4), 7.80 (1-H, d, J = 7.5 Hz, H-6), 7.50 (1H, t , J = 7.45 Hz, H-5), 3.54-3.78 (5H, m, H-7, H-8, H-9). 13C NMR (50 MHz, DMSO-d 6) δ: 197.9 (C=O), 139.3 (C-2), 134.8 (C-3), 134.2 (C-6), 128.4 (C-4), 128.2 (C-5), 60.4 (C-7, C-9), 53.5 (C-8). 11B NMR (128.4 MHz, D2O) δ: 7.3 ppm (h 1/2 = 256.8 Hz).
Compound 1g was synthesized from 0.05 g (0.304 mmol) of 4-acetylphenylboronic acid and 0.027 g (0.304 mmol) of serinol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.049 g (69 %); m.p. = 109-111°C. IR (ATR) ν (cm-1): 3089 (O-H, m), 2871 (N-H, w), 1674 (C=O, m), 1598 (w), 1541 (w), 1390 (w), 1372 (w), 1353 (w), 1278 (w), 1254 (w), 1186 (w), 1125 (w), 1050 (m), 957 (w), 861 (s), 827 (s), 800 (m), 747 (m), 629 (m). EI-MS m/z (%): 219 ([M-H2O]+,100), 204 (25), 187 (10), 172 (15), 161 (46), 120 (11), 100 (10). 1H NMR (200 MHz, D2O) δ: 7.88 (2H, d, J = 7.50 Hz, H-3, H-5), 7.68 (2H, d, J= 7.5 Hz, H-2, H-6), 3.62-4.02 (5H, m, H-7, H-8, H-9), 2.63 (3H, s, CH3). 13C NMR (50 MHz, D2O) δ: 204.9 (C=O), 134.9 (C-4), 132.0 (C-2, C-6), 127.4 (C-3, C-5), 60.0 (C-7, C-9), 53.8 (C-8), 26.0 (CH 3 CO). 11B NMR (128.4 MHz, D2O) δ: 2.0 ppm (h 1/2 = 513.6 Hz).
Compound 1h was synthesized from 0.05 g (0.304 mmol) of 3-acetylphenylboronic acid and 0.027 g (0.304 mmol) of serinol in 10 mL of MeOH. The product was obtained as a white powder. Yield 0.049 g (68 %); m.p. = 120-121 °C. IR (ATR) ν (cm-1): 3073 (O-H, m), 2875 (N-H, w), 1675 (C=O, m), 1510 (m), 1428 (w), 1392 (w), 1361 (w), 1268 (m), 1248 (m), 1148 (m), 1110 (m), 1043 (m), 989 (m), 873 (s), 821 (s), 737 (s), 622 (s). EI-MS m/z (%): 219 ([M-H2O]+, 100), 204 (63), 187 (30), 172 (48), 161 (94), 146 (17), 131 (14), 120 (31), 103 (24), 100 (34). 1H NMR (200 MHz, D2O) δ: 8.18 (1H, s, H-2), 7.84 (2H, m, H-4, H-6), 7.44 (1H, t, J= 7.7 Hz, H-5), 3.65-3.93 (5H, m, H-7, H-8, H-9), 2.67 (3H, s, CH3). 13C NMR (50 MHz, D2O) δ: 205.5 (C=O), 137.8 (C-2), 135.4 (C-3), 131.9 (C-6), 127.7 (C-4), 126.8 (C-5), 60.2 (C-7, C-9), 53.6 (C-8), 25.6 (CH 3 CO). 11B NMR (128.4 MHz, DMSO-d 6) δ: 1.8 ppm (h 1/2 = 924.5 Hz).
Compound 2e was synthesized from 0.184 g (1.23 mmol) of 4-formylphenylboronic acid and 0.112 g (1.23 mmol) of serinol in 30 mL of MeOH/Acetone (1:1 V/V) solvent mixture. Yield 0.104 g (32 %); m.p. = 103-105 °C. IR (ATR) ν (cm-1): 3103 (O-H, m), 2869 (N-H, w), 1682 (C=O, m), 1621 (C=C, m) 1556 (w), 1413 (w), 1318 (m), 1253 (s), 1151 (m), 1125 (m), 1047 (s), 1015 (s), 970 (s), 896 (s), 865 (s), 810 (s), 737 (s), 643 (s), 634 (s). EI-MS m/z (%): 245 ([M-H2O]+, 45), 230 (84), 187 (100), 145 (35), 129 (49), 77 (38), 60 (53). 1H NMR (200 MHz, DMSO-d 6) δ: 7.56-7.86 (5H, m, H-2,6, H-3,5, H-10), 6.83 (1H, d, 3 J trans = 16.5 Hz, H-11), 3.56-3.97 (5H, broad, overlapped with water signal, H-7, H-8, H-9), 2.32 (3H, s, H-13). 13C NMR (50 MHz, DMSO-d 6) δ: 198.2 (C=O), 144.7 (COCH), 134.6 (C-4), 133.4 (C-2, C-6), 132.3 (C-3, C-5), 127.2 (CH-Ar), 62.8 (C-7, C-9), 54.6 (C-8), 27.4 (CH 3 CO). 11B NMR (128.4 MHz, D2O) δ: 1.1 ppm (h 1/2 = 61.14 Hz).
Results and discussion
Zwitterionic boronate esters were prepared in a reaction of carbonylphenylboronic acid derivatives (4-formylphenylboronic acid; 1a, 1e), (3-formylphenylboronic acid; 1b, 1f), (4-acetylphenylboronic acid; 1c, 1g), 3-acetylphenylboronic acid; 1d, 1h) with 2-amino-2-methyl-1,3-propanediol (1a-1d) or serinol (1e-1h), in a 1:1 equimolar ratio using methanol as solvent (Scheme 1). The reaction mixtures were stirred at room temperature for 1 hour and upon standing over one night, a white precipitate appeared. The products were recovered by filtration and purified by crystallization from methanol resulting in moderate yields (~60 %). When the reaction of 4-formylphenylboronic acid with serinol in a 1:1 equimolar ratio was carried out using acetone/methanol as a solvent mixture (Scheme 2), compound 2e was obtained as a yellow pale solid in low yield (32 %). Besides the boronate ester formation, an α,β-unsaturated ketone was obtained as a product of the aldol condensation reaction involving the aldehyde group and the acetone solvent.


In all cases, the boronic acid reacts with the amino-diol and forms the boronic ester, and while the carbonyl groups were supposed to react with the amine group forming an imine bond, the boronic ester reacted with a water molecule forming a zwitterionic derivative. Thus, the amine group (NH2) is protonated yielding an ammonium group (NH3 +) and the hydroxyl group (-OH) is attached to the boron atom forming a boronate anion.
Spectroscopic analysis
All compounds 1a-1h and 2e were characterized by IR spectroscopy, mass spectrometry and 1H, 13C and 11B NMR spectroscopy. Furthermore, the structures of four zwitterionic boronate esters (1b, 1d, 1f and 2e) were established by single crystal X-ray diffraction. The first approximation to characterize the compounds was carried out by IR spectroscopy. In the IR spectra, all compounds 1a-1h and 2e showed broad absorption bands in the range from 3054 to 3105 cm-1 due to the presence of hydroxyl groups ν(OH), while the bands observed between 2849 to 2876 cm-1 correspond to the ammonium groups. Moreover, the band observed at 1666-1697 cm-1 was assigned to the stretching vibrations of the carbonyl group ν(C=O). Additionally, a new absorption band for compounds 1a-1h and 2e appear at 1043-1064 cm-1 which is characteristic of the stretching vibration of the ν(B-O) group. The formation of six-membered heterocyclic boronates 1a-1h was confirmed by EI-MS, where the spectra showed peaks at m/z = 219 (1a,1b), 233 (1c,1d), 205 (1e,1f), 219 (1i, 1h) and 245 (2e) corresponding to the molecular ion [M]+ with the loss of a water molecule. In most of the cases, this peak corresponds to the base peak indicating good stability of the compounds.
The characterization of compounds 1a-1h and 2e was complemented by NMR analyses. All compounds showed low solubility in common organic solvents. NMR spectra were recorded using DMSO-d 6 (1b, 1d and 2e) and D2O (1a, 1c, 1e, and 1h). The 1H NMR spectra of the formylboronic acid derivatives show signals at high frequency, between δ = 9.53-9.90 ppm assigned to the hydrogen atom of the aldehyde group. The signal for methylene hydrogens (CH2, H-7, H-9) for 2-amino-2-methyl-1,3-propanediol derivatives (1a-1d) was observed as an AB system at δ = 3.50-4.31 ppm, having coupling constants from 10.7 to 12.0 Hz. While for compounds 1e-1h and 2e, the signals corresponding to methylene hydrogens are observed as multiples and shifted between δ = 3.54-4.02 ppm. The 13C NMR also showed the corresponding signals for the carbonyl group which appears in the range of δ = 198.7-205.5 ppm for acetyl derivatives, while for aldehyde derivatives the signals were observed at a lower frequency (198.2-190.5 ppm). The carbon signals for aliphatic groups of the heterocyclic ring were observed between δ = 53.5-71.5 ppm. Finally, the 11B NMR spectra of all the compounds display a single broad signal in the range of δ = 1.9-7.3 ppm, as expected for tetracoordinated boron derivatives [35-39].
Suitable crystals for X-ray diffraction analysis were obtained for compounds 1b, 1d, 1f and 2e (Fig. 1) by recrystallization process from methanol solutions, crystallographic data are described at Table 1. Compounds 1d, 1f and 2e crystalized in monoclinic system, while 1b crystalized in a triclinic system. The asymmetric unit shows water molecules surrounding the boronate molecules, two for 1b, 1f, 2e, and four and a half for 1d.

Table 1 Selected crystallographic data for compounds 1b, 1d, 1f and 2e.
| 1b | 1d | 1f | 2e | |
|---|---|---|---|---|
| Empirical formula | C11H19BNO6 | C12H26BNO8.5 | C10 H17BNO6 | C13 H22BNO6 |
| Fw | 272.08 | 331.15 | 258.05 | 299.12 |
| Cryst size [mm3] | 0.15 x 0.24 x 0.32 | 0.22 x 0.27 x 0.45 | 0.23 x 0.36 x 0.40 | 0.22 x 0.36 x 0.44 |
| Cryst syst | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P1 | C2/c | P21/c | P21/c |
| Unit cell dimensions | ||||
| a [Å] | 6.5199(15) | 31.44(2) | 15.319(4) | 17.490(6) |
| b [Å] | 7.7679(18) | 9.062(6) | 12.176(4) | 12.586(4) |
| c [Å] | 14.562(3) | 12.104(8) | 6.908(2) | 6.842(2) |
| α [°] | 103.886(4) | 90 | 90 | 90 |
| β [°] | 90.806(4) | 94.275(10) | 96.023(5) | 94.147(6) |
| γ [°] | 103.850(4) | 90 | 90 | 90 |
| V [Å3] | 693.2(3) | 3439(4) | 1281.3(6) | 1502.1(9) |
| Z | 2 | 8 | 4 | 4 |
| ρ calcd [Mg/m3] | 1.304 | 1.279 | 1.338 | 1.323 |
| μ[mm-1] | 0.104 | 0.106 | 0.108 | 0.102 |
| Collected reflns | 2912 | 8993 | 11141 | 12307 |
| Independent reflns (R int) | 1892 (0.0275) | 2471 (0.0347) | 2001 (0.0834) | 2220 (0.1213) |
| Number of param | 187 | 240 | 190 | 209 |
| R1 [I > 2σ(I)] | 0.0751 | 0.0462 | 0.0497 | 0.0612 |
| wR2 (all data) | 0.1932 | 0.1257 | 0.1380 | 0.1557 |
| GOF | 1.083 | 1.119 | 1.074 | 0.900 |
Crystals for compounds 1a, 1c, 1e and 1g were also obtained (Fig. 2), however most of them include several disorder water molecules at the unit cell, for that reason, refinement of the structures was inadequate or included high R index values (Table S1). Nevertheless, the X-ray diffraction analysis allowed the observation of the whole molecular structures (only connective models).

The X-ray diffraction analysis shows a chair conformation for the six-membered heterocyclic ring for all eight compounds (Fig. 1). In fact, the presence of the -OH and -NH3 + groups at the six-membered ring gives the possible anti and syn isomers, resulting in four possible conformations (I-IV, Scheme 3). In this work, it was noticed, that the aryl and ammonium groups occupied the axial positions, whereas the methyl/proton and hydroxyl groups are in the equatorial positions. The above observation shows the preference for the syn stereoisomer considering the relationship between the hydroxyl and ammonium groups (isomer III at Scheme 3).

The endocyclic O-B bond distances are very similar (1.481(4) to 1.498(3) Å), being longer than the observed for the exocyclic B-O bond distances [1.453(3) -1.465(4) Å], values are related to similar reported boronate compounds [35-37, 49-51]. The angles observed around the boron atom are closer to the ideal value in a tetrahedral geometry (109.5°) having values of 104.1(4) to 113.9(4) º. In fact, the tetrahedral character (THC) for the boron atoms showed values of 80.5, 87.0 and 86.7 %, respectively [38,52], indicating the pseudo tetrahedral geometry (Table 2).
Table 2 Selected bond lengths (Å), angles (°) and THC for compounds 1b, 1d, 1f and 2e.
| Bonds (Å) | ||||
|---|---|---|---|---|
| Compound | 1b | 1d | 1f | 2e |
| B(1)-O(1) | 1.495(3) | 1.498(3) | 1.481(4) | 1.493(4) |
| B(1)-O(2) | 1.493(6) | 1.497(3) | 1.494(4) | 1.497(4 |
| B(1)-O(3) | 1.460(6) | 1.453(3) | 1.465(4) | 1.45(4) |
| B(1)-C Ar | 1.628(7) | 1.623(4) | 1.626(4) | 1.623(5) |
| Angles (°) | ||||
| O(3)-B(1)-O(2) | 108.8(4) | 106.8(2) | 107.7(2) | 1091(3) |
| O(3)-B(1)-O(1) | 104.1(4) | 107.06(19) | 108.6(2) | 1076(3) |
| O(2)-B(1)-O(1) | 106.6(4) | 108.56(19) | 108.1(2) | 1070(3) |
| O(3)-B(1)- C Ar | 112.2(4) | 110.5(2) | 108.0(2) | 1089(3) |
| O(2)-B(1)-C Ar | 110.9(4) | 112.5(2) | 112.3(2) | 1104(3) |
| O(1)-B(1)- C Ar | 113.9(4) | 111.1(2) | 113.0(2) | 113.8(3 |
| THC(%) | 80.5 | 87.0 | 86.7 | 88.2 |
As mentioned above, compounds 1a-1g crystallized with water molecules, which interact by hydrogen bonds with the boronates through the -OH and NH3 + groups. The supramolecular analysis for compounds 1b, 1d, and 1f in solid-state reveals the presence of several hydrogen bonds that result in the formation of 1D polymers. For instance, compound 1d is surrounded by seven water molecules via N-H···O and O-H···O interactions as depicted in Fig. 3(a). Moreover, it was observed the presence of six water molecules located between the donor and acceptor groups (-NH3 +, -OH), closing the interaction face to face of two boronate molecules as depicted in Fig. 3(b). The above observation favors the syn orientation of the OH and NH3 + groups as described previously at Scheme 3 (III). For compound 1d, the distances for the N-H···O donor-acceptor interactions have values of 1.962, 1.973 and 2.214 Å, whereas the O-H···O interaction has a distance of 2.014 Å. Fig. 3(c) and 3(d) show that the hydrogen bonds between the ammonium or hydroxyl groups with water molecules, support the formation of 1D polymer chains (Fig. S1), and give place to the generation of water channels along b-axis. Similar water chains were observed for compounds 1b and 1f (Figures S2 and S3) and analogous channels have been described for boron compounds [53] and hydrophilic organic molecules [54-56].
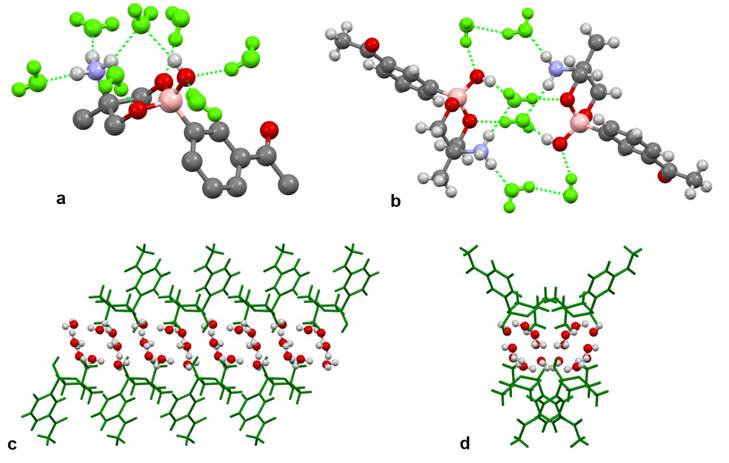
Fig. 3 (a) Seven water molecules have direct interaction with the boronate 1d via N-H···O and O-H···O hydrogen bonds. (b) Six water molecules connect face to face two boronate units. (c) Crystal packing for compound 1d along b-axis showing the polymeric chains of boronate and water molecules. (d) Crystal packing for compound 1d along a-axis.
Suitable crystals for the X-ray analysis were also obtained for compound 2e after filtration and slow evaporation of the mother solution. As mentioned above, complex 2e resulted from the aldol condensation reaction between the formyl group of 1e and the acetone solvent, giving rise to the formation of the respective carbon-carbon double bond in an E configuration. In fact, structural parameters related to the six-membered heterocycle conformation remain without significant changes, and bond distances and angles around the boron atom in complex 2e are comparable to its partners 1b, 1d, and 1f. The endocyclic B-O bond distances are slightly longer (1.493(4) -1.497(4) Å) compared with the exocyclic B-O distance (1.455(4) Å) of the hydroxyl group. The angles around the boron atom are between 107.0 to 113.8° with a THC value of 88.2 %, confirming the pseudo tetrahedral geometry. The -NH3 + and -OH groups preserve the syn conformation in the six-membered ring.
The supramolecular analysis for 2e reveals that each boronate molecule interacts with four water molecules via N-H···O and O-H···O hydrogen bonds (Fig. 4(a)). Furthermore, four water molecules connect two boronate units by hydrogen bonds (Fig. 4(b)). The distances for the N-H···O donor-acceptor interactions have values of 1.887, 1.986 and 2.090 Å, whereas the O-H···O interaction has a distance of 1.978 Å. It was noticed that the presence of (,(-unsaturated substituent does not affect the molecular packing, which looks like the observed for 1d. Once more, the observable 1D polymer chain for the boronate units results from the presence of water molecules connected via N-H···O and O-H···O hydrogen bonds (Figures 4(c) and S4).

Conclusion
Nine heterocyclic boron compounds were prepared by the condensation reaction of 2-amino-1,3-propanediol derivatives with carbonylphenylboronic acids at room temperature. Spectroscopic characterization indicated the formation of zwitterionic boronates, including the ammonium (NH3 +) and hydroxyl (OH) groups. The zwitterionic character decrease the solubility of compounds in common organic solvents, so, the possible reaction between the amino and carbonyl groups is inhibit, and a further self-assembly reaction was not achieved. Molecular structures obtained by X-ray diffraction analysis showed a chair conformation for the six-membered ring having the -NH3 + and -OH groups in a syn disposition. The presence of water molecules in the crystal network favored the formation of 1D polymeric chains supported by N-H···O and O-H···O hydrogen bonds. Using as solvent acetone, the aldol condensation was observed, while the boronate unit remains without modification; this reactivity opens the possibility to construct boronic acids with extending unsaturated moieties, which could be useful as sensors/indicators of anions, cations or neutral molecules.

