











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Montane cloud forests (MCF) cover 0.26% of the Earth’s surface, and less than 1% of the Mexican territory (Bubb et al., 2004). This ecosystem is characterized by a persistent cloud immersion (Rosas Rangel et al., 2019), occurring as patches at elevations of 600-3,200 m asl (Alfonso-Corrado et al., 2017; Ochoa-Ochoa et al., 2017; Santillán et al., 2020). It hosts a number of macroscopic endemic species (12% of the overall American mammal, bird and amphibian species; Hamilton, 2009; Karger et al., 2021), being recognized for its notorious levels of fungal diversity even at the small scale (Velez et al., 2021). The MCF provides vital ecosystemic services such as carbon capture, erosion control, as well as climate regulation, soil fertility, water supply and quality (Bazzaz, 1998; Bruijnzeel et al., 2010, 2011; Martínez et al., 2009). However, this unique biome ranks among the most threatened ecosystems globally, facing several stressors such as deforestation (Leija-Loredo & Pavón, 2017), climate change (Alfonso-Corrado et al., 2017), reduction of humidity (Santillán et al., 2020), increments in temperature (Foster, 2001), among others.
Microbial communities constitute important soil components (De Long et al., 2019) that fulfill key roles in edaphic nutrient cycles, serving as a sink and source of nutrients due to their remarkable ability to immobilize and release carbon (C), nitrogen (N), and phosphorus (P) in different chemical forms (Zak et al., 2003). This group includes several taxonomic assemblies (e.g., fungi, bacteria, virus, archaea and protists) of organisms smaller than 100µm (Wagg et al., 2018). Among these taxa, bacteria and fungi (hereafter referred to as a microbial assemblage, sensu Nemergut et al., 2013) are the largest and most diverse components comprising up to 90% of the overall microbial biomass in soils (Rinnan & Bååth, 2009). Hence, the understanding of this imperceptible, yet large component of soil diversity in MCFs represents a fundamental element for conservation.
Data from microcosm and field studies have demonstrated that microbial diversity and community composition influence soil ecosystem process rates (McGuire & Treseder, 2010). In this sense, bacteria and fungi collaborate in the decomposition and mineralization of organic remains (Romaní et al., 2006; Tapia-Torres & García-Oliva, 2013), driving the development of edaphic stable and labile pools of C, N and other nutrients, which facilitate the subsequent establishment of plant communities (Schulz et al., 2013). In forest systems, bacteria carry out the hydrolysis and mineralization of organic matter through the biosynthesis of exoenzymes, followed by the release and uptake of nutrients (PO4 - and NH4 +) from the soil solution. Emblematic taxa with these capacities include members of Pseudomonas, Burkholderia, Escherichia, Serratia, Bacillus, Enterobacter, Nostoc, Caulobacter, Sinorhizobium, Mesorhizobium, and Corynebacterium (Horwath, 2017; Idriss et al., 2002).
Additionally, edaphic fungi perform several ecological roles as pathogens, saprotrophs, and symbionts (Nguyen et al., 2016). These osmotrophs play essential roles in nutrients turnover (Zanne et al., 2020), depolymerizing recalcitrant lignin and cellulose molecules contained in leaf and wood litter through the production of extracellular enzymes (de Boer et al., 2005). Furthermore, fungal pathogenic taxa act as biological control agents, being implicated in plant diversity maintenance (Brown et al., 2011). To the best of our knowledge, soil microfungal diversity in Mexican MCFs includes members affiliated to Alternaria, Aspergillus, Bipolaris, Chaetomium, Cladosporium, Cordana, Curvularia, Chalara, Dictyochaeta, Fusarium, Gyrothrix, Humicola, Monodictys, Myrmecridium, Penicillium, Periconia, Pestalotiopsis, Sporidesmium, Stachybotrys, Talaromyces , Trichoderma, and Virgaria (Arias & Heredia-Abarca, 2014, 2020; Heredia-Abarca et al., 2011). Also, several ectomycorrhizal fungi have been linked with roots of Juglandaceae species (Corrales et al., 2021).
The generation and amalgamation of diversity data at different scales is fundamental to develop a broad understanding of ecosystems (Oda et al., 2019). At the large scale, MCFs have been extensively investigated, reporting high heterogeneity and diversity levels (Williams et al., 2013). Though, small-scale studies have received less attention with respect to larger macroscale explorations. Pioneer efforts analyzing biogeochemical data have demonstrated an environmentally heterogeneous setting, with enzymatic activities suggesting distinctive small-scale soil patterns (Velez et al., 2021). Nevertheless, microbial diversity patterns remain poorly understood at the small scale in this environment, hampering the robust view of ecosystem functioning as small-scale processes may be masked by larger scale features (Mori et al., 2018).
In view of MCFs vulnerability to anthropogenic stressors, and the lack of knowledge on soil microbial diversity and its relationship with ecosystem processes (e.g., nutrient cycling) at different scales, herein we evaluated cultivable soil microbial diversity and community structure associated with the soil below 2 iconic plant taxa (endemic, relict, and endangered species) in a pristine location of Mexican MCF at the small spatial scale. We predict that our approach will lead to the predominant isolation of saprotrophic fungi and potentially phosphate solubilizer bacteria; in addition, we hypothesize that the small-scale distribution of microbial assemblages will be strongly associated with environmental variables such as soil phosphorous availability.
Materials and methods
The fieldwork was conducted in the MCF locality of El Relámpago (17°35’30.4” N, 96° 23’57.1” W; at 2,219 m asl), within the municipality of Santiago Comaltepec, in the mountainous system of northern Oaxaca (del Mar Delgado-Serrano et al., 2015). This forest harbors high numbers of endemic vertebrate species and a genetically diverse population of Oreomunnea mexicana, due to its good conservation status (del Mar Delgado-Serrano et al., 2015; Ponce-Reyes et al., 2012, 2020). The climate is usually temperate-humid with rainfall in summer (INEGI, 2010), an annual average temperature of 16-20 °C, and average annual precipitation of 2,000-4,500 mm (Trejo, 2004). The main soil type is Acrisol, which is strongly acidic, with a subsurface horizon of clay accumulation and low nutrient retention capacity (Alfaro-Sánchez, 2004; Krasilnikov et al., 2013). Velez et al. (2021) described the biogeochemical characteristics of the study site, highlighting high edaphic heterogeneity at the small spatial scale, abundant total carbon (TC) and dissolved organic carbon (DOC), as well as high polyphenol oxidase (POX) activity values.
We employed a triangular sampling method as proposed by Bąk (2014) . Therefore, 3 sampling plots were set up along a 10 m-triangular transect, considering different elements of MCF for a greater representation of the microbial community: the first plot was established adjacent to an individual of O. mexicana (17°35’0.36” N, 96°23’36.6” W), a representative and endangered species of Mexican MCF (Alfonso-Corrado et al., 2017; Rzedowski, 1996); the second plot was settled next to an individual of Alsophila salvinii (17°35’18.4” N, 96°23’57.7” W), a conspicuous fern in the region (Rzedowski & Palacios-Chávez, 1977); and the third plot corresponded to the area under a fallen wooden log (17°34’51.83” N, 96°24’5.17” W). Subsequently, in each plot a 1 m-equilateral triangular subplot was traced (Fig. 1). In total, 9 soil cores (3 per plot) were sampled in the first 10 cm of soil (excluding litter) using sterile Falcon tubes and transported in a cooler to the laboratory within the next 48 h for processing.
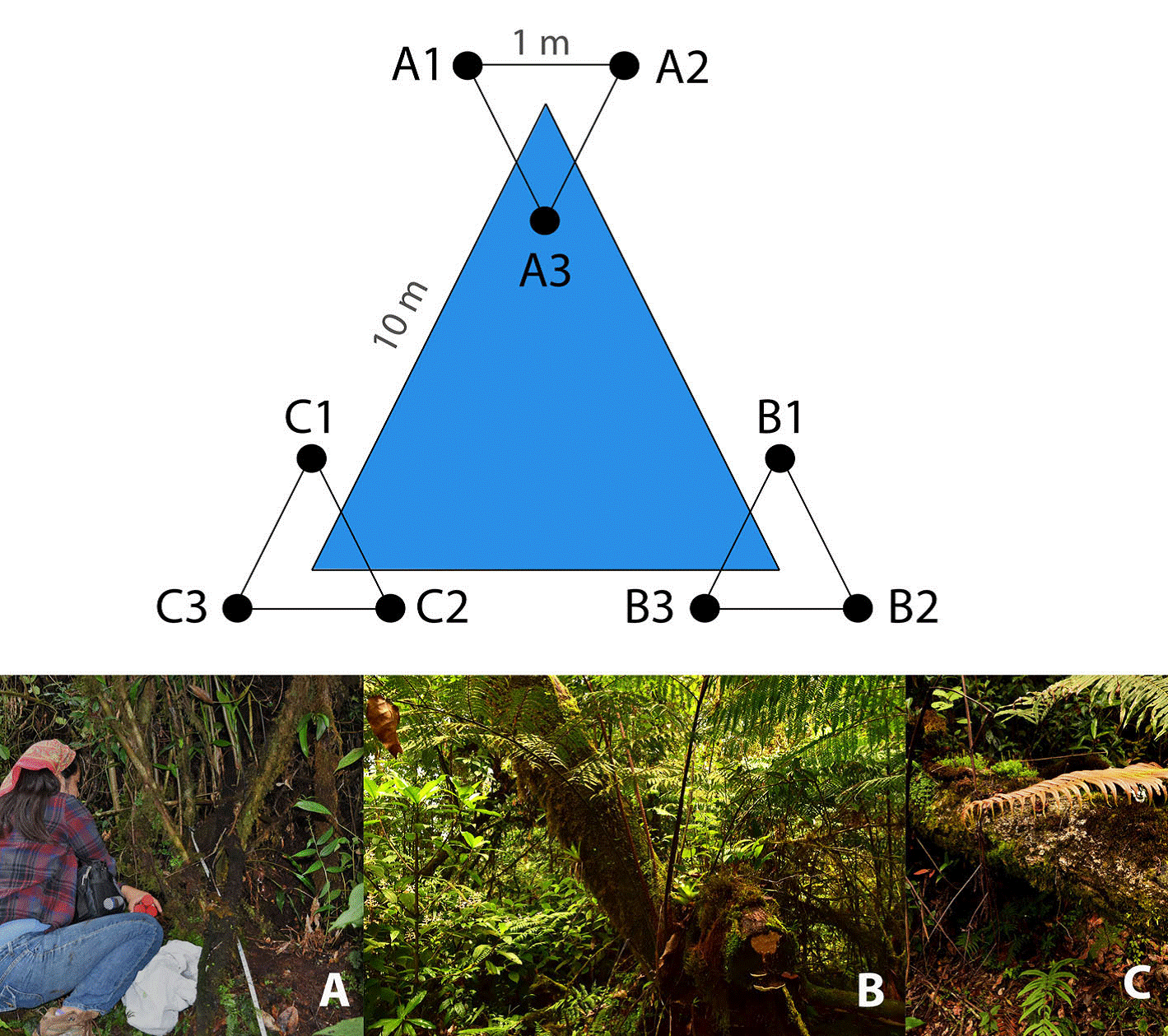
Figure 1 Triangular sampling design where 3 plots were established in: (A) at the base of Oreomunnea mexicana, (B) at the base of Alsophila salvinii, and (C) at the base of a fallen decaying tree; subsequently in each plot a 1 m-equilateral triangular subplot was traced for soil sampling.
During the fieldwork we detected a visibly sick population of A. salvinii (presence of dark spots and blights on fronds), including the individual within our sampling plot (Fig. 2). So, despite that this was not part of the objectives of this study, and given the importance of prompt disease detection in threatened ecosystems in order to mitigate outbreaks, samples (consisting of sick fronds individually placed in Zip-lock® plastic bags) were collected to characterize the etiological agent. All the material was immediately stored and transported at 4º C in the dark to the laboratory and processed within the next 48 h.

Figure 2 Fronds of Alsophila salvinii showing disease symptoms such as the presence of dark spots and blights, indicated by the arrow.
Fungi and bacteria were isolated using the dilution plating method (Warcup, 1960) on Potato Dextrose Agar (PDA; Fluka Analytical, Sigma-Aldrich), Corn Meal Agar (CMA; Fluka Analytical, Sigma-Aldrich), Luria Bertani Agar (LB; Lennox Agar, Invitrogen), and LB− (2-fold diluted LB). Dilution plates were prepared using 1 g of soil sample, at 10-1 - 10-6 dilutions in test tubes with sterilized distilled water. Three replicates for each dilution of each sample were plated (1 ml aliquot). Petri dishes were incubated at laboratory room temperature (22-25 °C), with a 12 h photoperiod, and examined periodically for up to 2 weeks. During this period, different colony morphologies from each medium were transferred and maintained on PDA and LB plates for fungi and bacteria respectively.
Fronds from A. salvinii were initially washed in running tap water. Next, surface sterilization was attained by sequential solutions of 70% ethanol (1 min), 2.6% sodium hypochlorite (3 min), and 70% ethanol (1 min). Small pieces (0.5 × 0.5 cm) from sections including dark spots and blights on leaves were placed in Petri dishes containing PDA, and incubated at 25 °C for 10 to 15 days. Following incubation, fungal axenic isolates were obtained and subsequently transferred to PDA for maintenance.
Fungi were identified by evaluating morphological characteristics, combined with the analysis of the ITS1-5.8S-ITS2 rDNA, hereafter referred to as the ITS region.
So, genomic DNA of the axenic isolates was extracted using the protocol described by Doyle and Doyle (1987) . The ITS region was amplified by using primers set ITS1 and ITS4 as reported by White et al. (1990) . The bacterial genomic DNA from axenic cultures was isolated using Dneasy Blood & Tissue Kit®. The 16S ribosomal DNA region was amplified with primers 27F and 1492R (Lane, 1991). The PCR products were sequenced in both directions using a 3730xl DNA Analyzer (Applied Biosystems™) at LANABIO, Biology Institute, National Autonomous University of Mexico (UNAM). Cultures and total DNA are stored in the culture collection of the Laboratory C-121, Biology Institute, UNAM, headed by Dr. Patricia Velez, and are fully available for research upon request.
Quality assessment and assembly of the ITS region and 16S Sanger sequences from fungal and bacterial isolates was performed using the finishing tool Consed version 29.0 (Ewing & Green, 1998; Ewing et al., 1998; Gordon et al., 2001). For the taxonomic assignment, sequence homology was evaluated through the comparison against the UNITE database for fungi (Kõljalg et al., 2020; Nilsson et al., 2019), and sequences from type material of the National Center of Biotechnology Information GenBank database using the BLAST algorithm for bacteria through a BLAST search (Abarenkov et al., 2010; Kõljalg et al., 2013). Sequence similarity for defining OTUs was set with a cut-off value of 98-100% for presumed species, 94-97% for genus level and 80-93% for order level. For conflicting hits, the lowest common rank level was used (Peršoh et al., 2010; Table 1). The sequences were deposited in GenBank under the accession numbers MT108978-MT109012 for fungi (Table 1), and MZ048754-MZ048770 for bacteria (Table 2).
Table 1 Fungal isolates obtained from soil samples collected in a pristine location of Mexican cloud forest. * Isolated from sick fronds of A. salvinii.
| Isolate | ITS1-5.8S-ITS2 | ||||
| OTU | Reference Accession numbers | % Identity | e-value | Accession NCBI | |
| N10 | Aspergillus inflatus | MH859900 | |||
| MH859521 | |||||
| MH859519 | |||||
| AJ608959 | 99 | 0 | MT108998 | ||
| AF033393 | |||||
| T8 | Aureobasidium pullulans | MT882127 | |||
| MH864403 | |||||
| JX188099 | 100 | 0 | MT109010 | ||
| EU272483 | |||||
| MF062189 | |||||
| M22_T13 | Beauveria sp. | AY532003 | |||
| HQ880820 | |||||
| MH865206 | 99 | 0 | MT108987 | ||
| MH862139 | |||||
| HQ880819 | |||||
| N3_T2_T4_T9_M6 | Cladosporium sp. | MN543985 | |||
| MN543962 | |||||
| MN543951 | 100 | 0 | MT109002 | ||
| MN521809 | |||||
| MN518420 | |||||
| M5_2A | Clavicipitaceae sp. | MN905773.1 | |||
| HM030580 | |||||
| MH864652.1 | 100 | 0 | MT108996 | ||
| MH859547.1 | |||||
| AB709835.1 | |||||
| M19_Tube 12 | Clonostachys rosea* | MN511326 | |||
| KX421414 | |||||
| HM052817 | 99 | 0 | MT109000 | ||
| KM265525 | |||||
| MH859090 | |||||
| T19_N16 | Diaporthaceae sp. | MH864503.1 | 99 | 0 | MT108997 |
| MH299960.1 | |||||
| MH299958.1 | |||||
| MH020798.1 | |||||
| FN597586.1 | |||||
| M20_M26 | Diaporthe sp. | MF435154 | |||
| MF435146 | |||||
| MF435133 | 100 | 0 | MT108986 | ||
| MF435132 | |||||
| MF435131 | |||||
| M10_N11_M28 | Didymellaceae sp. | MH861244 | |||
| MN077427 | |||||
| JF817335 | 99 | 0 | MT108978 | ||
| JN207257 | |||||
| MF435134 | |||||
| M23 | Dothidiomycetes sp. | MN421894 | |||
| MN421889 | |||||
| MN421869 | 99 | 0 | MT108981 | ||
| KX640595 | |||||
| KX640594 | |||||
| M13 | Furcasterigmium furcatum | MH859660 | |||
| MH856099 | |||||
| AJ608973 | 99 | 0 | MT108980 | ||
| JF311914 | |||||
| LR590130 | |||||
| M2B | Fusariella sp. | EU687056 | 96 | ||
| KF800481 | 94 | ||||
| FJ820737 | 94 | 0 | MT108986 | ||
| MH859784 | 94 | ||||
| MH860688 | 93 | ||||
| M25 | Gaeumannomyces californicus | NR_155135.1 | 98.149 | ||
| NR_155133.1 | 97.799 | ||||
| KX306490.1 | 98.149 | 0 | MT108990 | ||
| KX306480.1 | 97.799 | ||||
| KX306482.1 | 97.269 | ||||
| T14 | Ilyonectria sp. | KP761750 | 100 | 0 | MT109005 |
| MK164179 | |||||
| KF895008 | |||||
| MF101382 | |||||
| LC133803 | |||||
| Tube 8_Tube 9 | Mariannaea sp. | MH863675 | |||
| MH862153 | |||||
| KM231758 | 100 | 0 | MT109001 | ||
| KM231757 | |||||
| KF767354 | |||||
| N14 | Metarhizium anisopliae | MH864642 | |||
| MH483803 | |||||
| KY786031 | 99 | 0 | MT109000 | ||
| EU307915 | |||||
| AF137059 | |||||
| M34 | Metarhizium carneum | MK164228 | |||
| MK164227 | |||||
| HQ392598 | 98 | 0 | MT109012 | ||
| MK387968 | |||||
| EU553292 | |||||
| M9 | Mortierellaceae sp. | MH860437 | |||
| MH860436 | |||||
| MH860435 | 99 | 0 | MT108997 | ||
| MH860122 | |||||
| MH860121 | |||||
| M17 | Mortierella turficola | EF521229 | |||
| AM292200 | |||||
| EU240043 | 99 | 0 | MT108982 | ||
| JX976025 | |||||
| JX975952 | |||||
| T11 | Nectriaceae sp. | KP265346 | 97 | ||
| MT534189 | 96 | ||||
| HQ897787 | 96 | 0 | MT109004 | ||
| HQ897787 | 96 | ||||
| AM410602 | 96 | ||||
| N13 | Parapyrenochaeta acaciae | KX228265 | 100 | ||
| KF673765 | 99 | ||||
| MK441755 | 95 | 0 | MT108999 | ||
| KX147607 | 98 | ||||
| KX147606 | 98 | ||||
| M14 | Parengyodontium album | MK834516 | 99 | 0 | MT109008 |
| MW187752 | |||||
| MW077094 | |||||
| MT672589 | |||||
| MT626052 | |||||
| M1A_M1B | Parengyodontium album | MK719933 | |||
| MH860372 | |||||
| LC092885 | 100 | 0 | MT108985 | ||
| LC092884 | |||||
| LC092882 | |||||
| N30_T5 | Penicillium sp. | NR_077153 | |||
| MN515068 | |||||
| MN511336 | 100 | 0 | MT109003 | ||
| MN371392 | |||||
| MT872087 | |||||
| M18_M3 | Phomopsis sp. | MF185326 | |||
| EU002915 | |||||
| MF185359 | 99 | 0 | MT108983 | ||
| MF185341 | |||||
| MF185334 | |||||
| T22 | Pleosporales sp. 1 | FM178244 | 99 | ||
| FM178246 | 98 | ||||
| MK066907 | 99 | 0 | MT109008 | ||
| MH931265 | 99 | ||||
| MH844084 | 99 | ||||
| M24 | Pleosporales sp. 2 | MH935005 | 100 | ||
| KY367514 | 99 | ||||
| KT309810 | 100 | 0 | MT108989 | ||
| MH861839 | 99 | ||||
| KY940787 | 99 | ||||
| Tube 6 | Pleosporomycetidae sp. | KJ591760 | 96 | ||
| KY454761 | 96 | ||||
| LT623218 | 96 | 0 | MT108992 | ||
| KF811432 | 95 | ||||
| JQ388267 | 95 | ||||
| M11 | Setophaeosphaeria | KJ869161 | 99 | ||
| hemerocallidis | |||||
| KX515692 | 97 | ||||
| KX515688 | 97 | 0 | MT108979 | ||
| KX515679 | 97 | ||||
| KX515674 | 97 | ||||
| M28 | Talaromyces wortmannii | MK020174 | 100 | 0 | MT108991 |
| KF984826 | |||||
| KF984825 | |||||
| KF984824 | |||||
| KF984823 | |||||
| M35_M27_M30_ | Tolypocladium geodes | MH859919 | |||
| M33_T20_M15_ | |||||
| M32 | |||||
| KU556539 | 99 | 0 | MT108995 | ||
| JX507694 | |||||
| T16B_T17 | Trichoderma sp. 1 | MN516473 | |||
| MN516472 | |||||
| MN186861 | 100 | 0 | MT109006 | ||
| MN186859 | |||||
| MK871069 | |||||
| Tube 4 | Trichoderma sp. 2 | MN518401 | |||
| MN516457 | |||||
| MN516456 | 100 | 0 | MT109011 | ||
| MN516454 | |||||
| MN516452 | |||||
| T6 | Trichoderma koningii | Z79628 | |||
| X93983 | |||||
| MN516479 | 100 | 0 | MT109010 | ||
| MN516476 | |||||
| MN516475 | |||||
| M29 | Wojnowiciella dactylidis | LT990661 | |||
| LT990659 | |||||
| LT990658 | 99 | 0 | MT108993 | ||
| MK442631 | |||||
| KF800363 |
Table 2 Bacterial isolates obtained from soil samples collected in a pristine location of Mexican cloud forest.
| Isolate | 16S | ||||
| OTU | Accession NCBI | % Identity | e-value | Accession NCBI | |
| 28.P | Arthrobacter sp. | MW227493.1 | 100 | ||
| NR_133969.1 | 98.45 | 0 | MZ048754 | ||
| JX949648.2 | 98.01 | ||||
| MN080869.1 | 98.01 | ||||
| NR_170399.1 | 98.01 | ||||
| C4 | Bacillus sp. | CP009692.1 | 100 | ||
| NR_113990.1 | 100 | ||||
| AM747229.1 | 100 | 0 | MZ048770 | ||
| NR_115993.1 | 100 | ||||
| NR_036880.1 | 100 | ||||
| N10 | Microbacterium sp. | MK424288.1 | 100 | ||
| NR_042263.1 | 100 | ||||
| MT760166.1 | 99.47 | 0 | MZ048756 | ||
| NR_117603.1 | 99.47 | ||||
| MT760185.1 | 97.87 | ||||
| 47.P | Planococcaceae sp. 1 | NR_113837.1 | 99.78 | ||
| NR_029233.1 | 99.78 | ||||
| X68415.1 | 99.78 | 0 | MZ048755 | ||
| NR_113752.1 | 99.57 | ||||
| MT760068.1 | 99.57 | ||||
| N12 | Planococcaceae sp. 2 | NR_025628.1 | 98.22 | 1.00E-165 | |
| NR_025627.1 | 98.22 | 1.00E-165 | MZ048757 | ||
| NR_109749.1 | 97.63 | 2.00E-162 | |||
| NR_041521.1 | 97.63 | 2.00E-162 | |||
| NR_118296.1 | 97.33 | 1.00E-160 | |||
| N13 | Planococcaceae sp. 3 | NR_025627.1 | 97.93 | 0 | |
| NR_025628.1 | 97.67 | 0 | MZ048758 | ||
| NR_025029.1 | 95.61 | 4.00E-175 | |||
| NR_041521.1 | 95.09 | 9.00E-172 | |||
| NR_144702.1 | 94.07 | 2.00E-164 | |||
| 46.P1 | Planococcaceae sp. 4 | NR_116601.1 | 97.86 | ||
| CP016539.2 | 97.69 | 0 | MZ048763 | ||
| CP013659.2 | 97.69 | ||||
| LC379145.1 | 97.69 | ||||
| NR_113814.1 | 97.69 | ||||
| 50.P2 | Planococcaceae sp. 5 | KU886574.1 | 94.93 | ||
| NR_171442.1 | 94.93 | 0 | MZ048764 | ||
| NR_134133.1 | 94.8 | ||||
| CP016534.2 | 94.74 | ||||
| CP016539.2 | 94.75 | ||||
| T2 | Planococcaceae sp. 6 | NR_113752.1 | 99.77 | ||
| MT760068.1 | 99.77 | 0 | MZ048769 | ||
| MT757992.1 | 99.77 | ||||
| NR_113837.1 | 99.08 | ||||
| NR_036942.1 | 99.08 | ||||
| T11 | Pseudomonas sp. 1 | LT629778.1 | 100 | ||
| CP062253.1 | 99.69 | 0 | MZ048759 | ||
| CP029608.1 | 99.54 | ||||
| KT321658.1 | 99.54 | ||||
| CP062252.1 | 99.54 | ||||
| 29.P1 | Pseudomonas sp. 2 | MT027239.1 | |||
| NR_103934.2 | 99.66 | 0 | MZ048760 | ||
| NR_148295.1 | |||||
| NR_134795.1 | |||||
| LK021121.2 | |||||
| N8 | Pseudomonas sp. 3 | NR_148295.1 | 99 | ||
| MW111151.1 | 98.67 | 0 | MZ048761 | ||
| LR134290.1 | 98.01 | ||||
| LC507444.1 | 97.84 | ||||
| NR_134795.1 | 97.84 | ||||
| 40.P | Pseudomonas sp. 4 | LT629790.1 | 99.32 | ||
| LC409077.1 | 99.01 | 0 | MZ048762 | ||
| MZ099645.1 | 99.01 | ||||
| LC409075.1 | 98.94 | ||||
| NR_025102.1 | 98.86 | ||||
| C1 | Pseudomonas sp. 5 | JX545210.1 | 99.51 | ||
| LC595308.1 | 99.51 | 0 | MZ048767 | ||
| MK680061.1 | 99.18 | ||||
| MG719526.1 | 99.18 | ||||
| CP009533.1 | 99.18 | ||||
| C2 | Pseudomonas sp. 6 | LC500864.1 | 100 | ||
| LC548100.1 | 99.86 | 0 | MZ048768 | ||
| KX186943.1 | 99.86 | ||||
| KX186942.1 | 99.86 | ||||
| KX186936.1 | 99.86 | ||||
| N251 | Xanthomonadaceae sp. | NR_121739.1 | 98.73 | ||
| CP007597.1 | 98.73 | 0 | MZ048765 | ||
| MW629800.1 | 98.73 | ||||
| KY020782.1 | 98.36 | ||||
| NR_028930.1 | 98.37 | ||||
| T17 | Xanthomonadaceae sp. | NR_121739.1 | 99.47 | ||
| CP007597.1 | 99.47 | 0 | MZ048766 | ||
| MW629800.1 | 99.47 | ||||
| NR_028930.1 | 99.47 | ||||
| AJ293463.1 | 99.47 | ||||
Statistical analyses
We evaluated the relationship between microbial community structure and the following biogeochemical data retrieved from Velez et al. (2021; synchronously collected from the exact same sampling sites): 1) soil physicochemical properties: pH, NH4 +, TC, total nitrogen and phosphorus (TN and TP respectively), DOC, dissolved nitrogen and phosphorus (DON and DOP respectively) and forms of carbon, nitrogen, and phosphorus contained in microbial biomass (Cmic, Nmic and Pmic respectively), and 2) 6 soil exoenzymes: β-1,4-glucosidase (BG), cellobiohydrolase (CBH), β-1,4-N-acetylglucosaminidase (NAG), phosphomonoesterase (AP), phosphodiesterase (APD), and POX. The raw data matrix was normalized using Z scores. We evaluated clustering patterns among sampling sites based on the culturable microbial community and environmental variables with the “hclust” function in ade4 v1.7-13 package in R (Dray & Dufour, 2007). A Principal Component Analysis (PCA) and a Spearman correlation test between soil biogeochemical variables and enzyme activities were conducted to select the variables for the subsequent multivariate analysis aiming to elucidate the relationships between biological assemblages of species and their environment. For the PCA, we considered as informative the components that represented at least 85% of the accumulative variance; whereas for the Spearman correlation matrix, we defined an uncorrelated model by using a threshold of 0.85 (Booth et al., 1994). These analyses were computed in R software 3.6.0 (R Core Team, 2018) using FactoMineR version 2.1 (Lê et al., 2008). Next, a Canonical Correspondence Analysis (CCA) with the selected biogeochemical variables (pH, DOC, DON, DOP, NH4+, POX, NAG and AP) and species data was calculated using the R package vegan (Oksanen et al., 2009).
Results
Overall 101 axenic fungal isolates were obtained from the 9 soil subsamples, clustering into 35 OTUs. The OTUs belonged to the phyla Mortierellomycota (Mortierellaceae sp. and Mortierella turficola), and Ascomycota (33 OTUs). The Ascomycota represented the most abundant and diverse phylum in our samples; affiliated with 8 orders: Capnodiales (1 OTU), Diaporthales (3 OTUs), Dothideales (1 OTU), Eurotiales (3 OTUs), Glomerellales (1 OTU), Hypocreales (14 OTUs), Magnaporthales (1 OTU) and Pleosporales (6 OTUs). At the genus level, isolates of the Ascomycota belonged to 21 genera: Aspergillus, Aureobasidium, Beauveria, Cladosporium, Clonostachys, Diaporthe, Furcasterigmium, Fusariella, Gaeumannomyces, Ilyonectria, Mariannaea, Metarhizium, Parapyrenochaeta, Parengyodontium, Penicillium, Phomopsis, Setophaeosphaeria, Talaromyces, Tolypocladium, Trichoderma and Wojnowiciella (Table 1). Among these, the dominant component was Tolypocladium geodes.
We isolated 170 bacterial strains out of which representative isolates with distinctly unique morphologies were identified based on the homology of the 16S rRNA gene region towards reference sequences from the NCBI database. A total of 17 OTUs were delimited within the Actinobacteria (Arthrobacter and Microbacterium), Firmicutes (Bacillus) and Proteobacteria (Pseudomonas). The most abundant elements were Pseudomonas and Bacillus representatives.
The distance dendrogram showed a sparse clustering among sampling sites (Fig. 3). Likewise, the PCA confirmed a considerable heterogeneity in the soil environmental data, with the first 2 ordination axes explaining 61.81% of the total variation (Fig. 4). The variables that most contributed to the first component were TC, TN, AP, NH4 + and TP; whereas for the second component BG, DON, Pmic and DOP showed the top contribution (Fig. 5). Spearman analysis showed significant correlations among biogeochemical variables, such as: NH4 +, DOC and DOP, as well as AP, NAG, APD and POX (Supplementary material, Tables 1, 2).
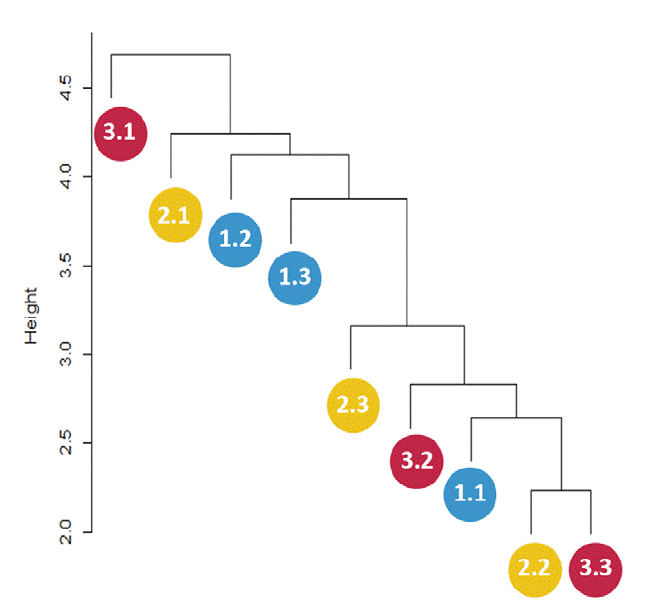
Figure 3 Distance dendrogram showing sparse clustering patterns among sampling sites based on the culturable microbial community. Blue circles represent sampling subplots at the base of Oreomunnea mexicana (1.1, 1.2, and 1.3), yellow circles represent sampling subplots at the base of Alsophila salvinii (2.1, 2.2, and 2.3), and red circles represent sampling subplots at the base of a fallen decaying tree (3.1, 3.2, and 3.3).
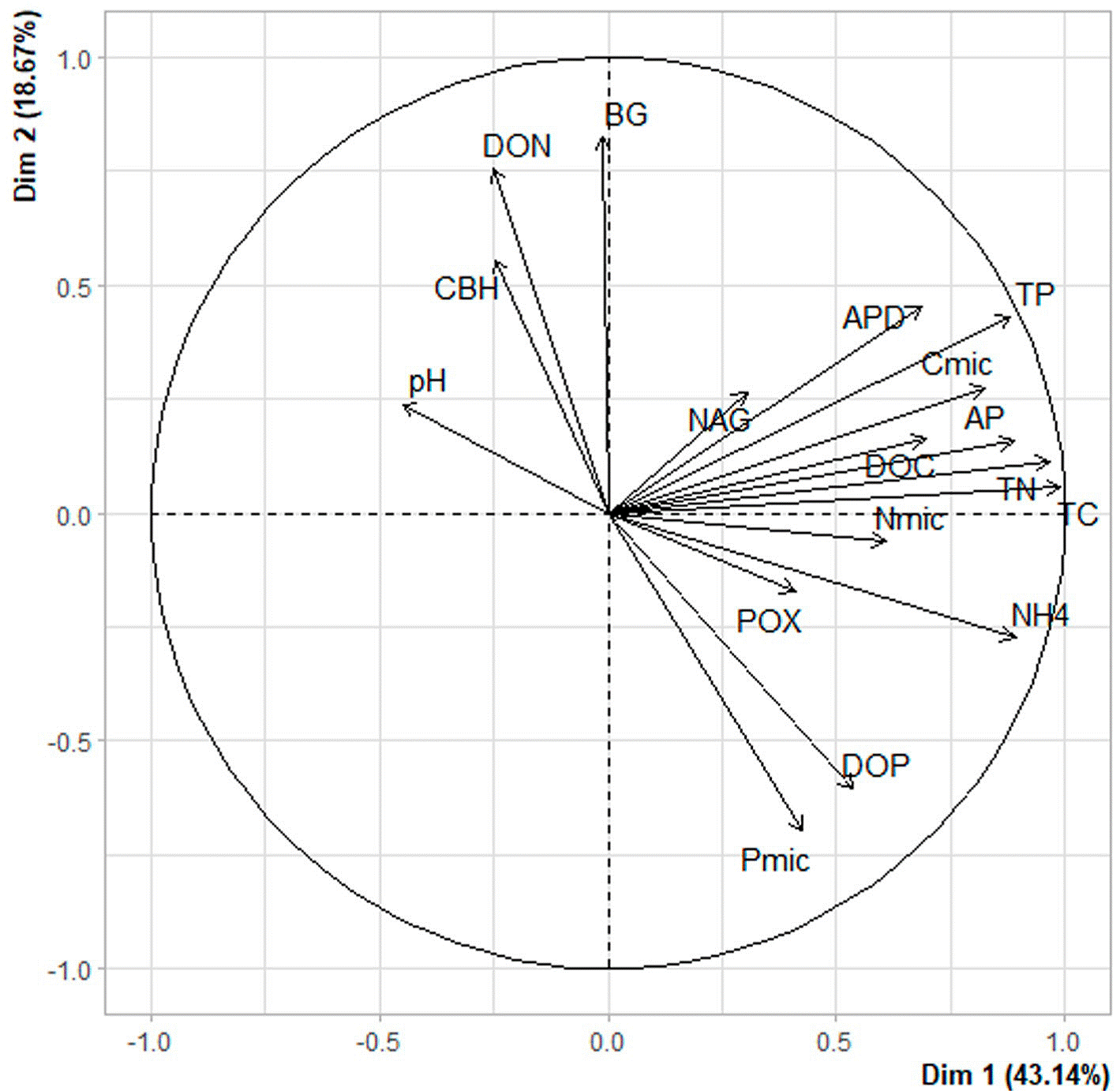
Figure 4 Principal component analysis of environmental variables published in Vélez et al. (2021), including: β-1,4-glucosidase (BG); cellobiohydrolase (CBH); β-1,4-N-acetylglucosaminidase (NAG); phosphomonoesterase (AP); phosphodiesterase (APD); polyphenol oxidase (POX); ammonium (NH4 +); TC, TN and TP are total carbon, nitrogen and phosphorus; DOC, DON and DOP are dissolved organic carbon, nitrogen and phosphorus; Cmic, Nmic and Pmic are carbon, nitrogen and phosphorus in microbial biomass.
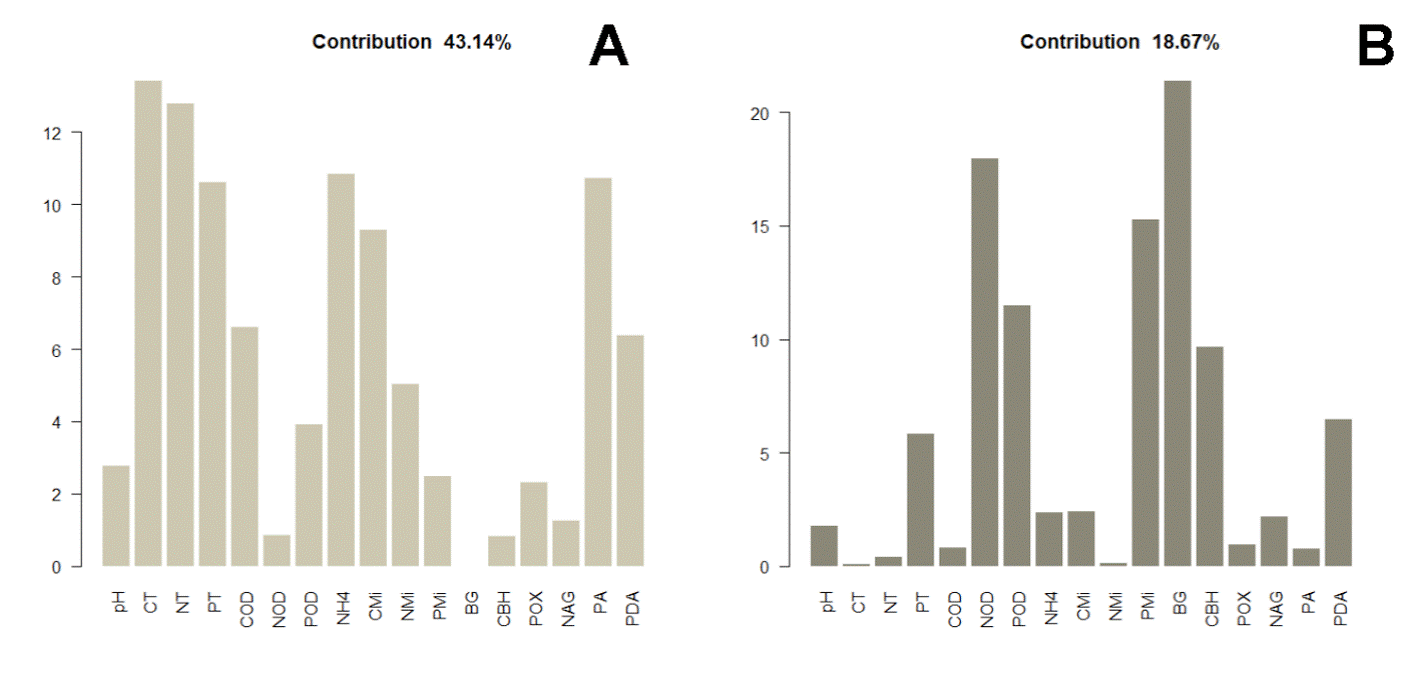
Figure 5 Individual contribution of variables to the first (A) and second component (B) of the PCA, where β-1,4-glucosidase (BG); cellobiohydrolase (CBH); β-1,4-N-acetylglucosaminidase (NAG); phosphomonoesterase (AP); phosphodiesterase (APD); polyphenol oxidase (POX); ammonium (NH4 +); TC, TN, and TP are total carbon, nitrogen and phosphorus; DOC, DON, and DOP are dissolved organic carbon, nitrogen and phosphorus; Cmic, Nmic, and Pmic are carbon, nitrogen and phosphorus in microbial biomass.
The CCA data suggested that the distribution of fungal and bacterial assemblages in the soil is strongly associated with key environmental variables (Fig. 6). For instance, we detected 5 relevant associations: 1) T. geodes and DON; 2) Penicillium sp., Diaporthaceae sp., and DOC; 3) Pleosporales sp. M24, Xanthomonadaceae sp. N251, and POX; and 4) M. turficola, Dothideomycetes sp., Metarhizium carneum, Wojnowiciella dactylidis, and NH4 + (linked to the samples collected near the fern A. salvinii).
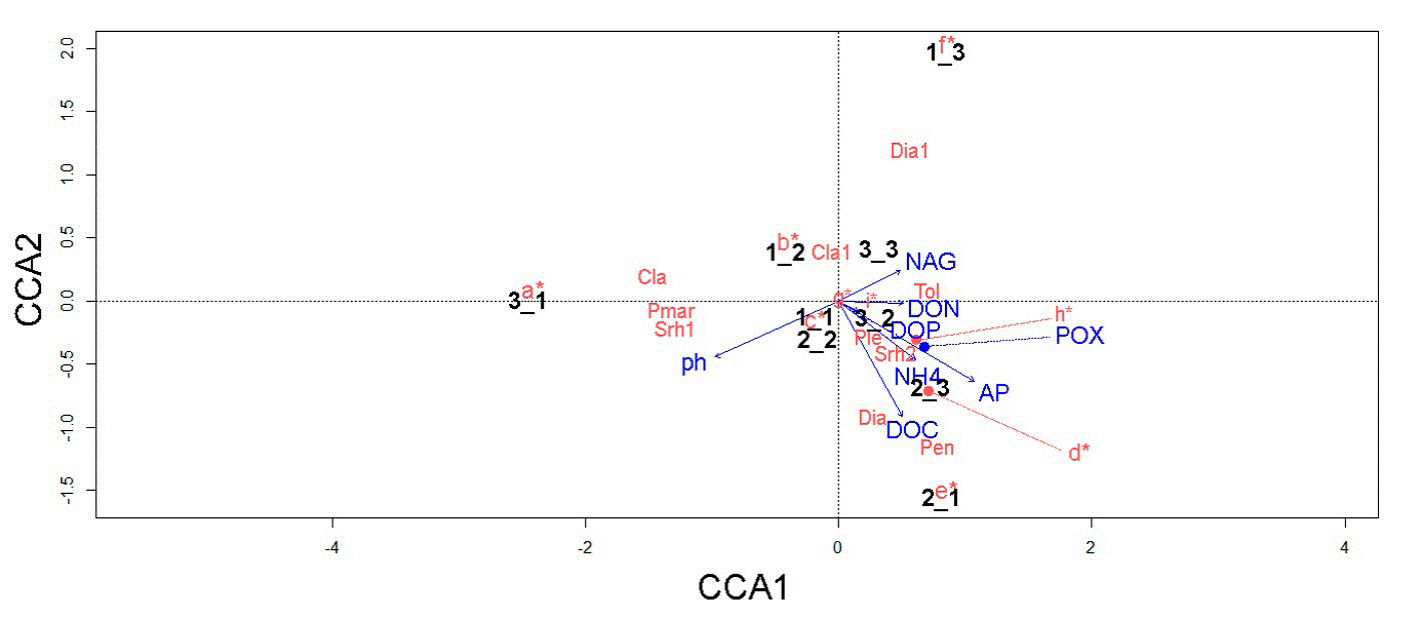
Figure 6 Canonical correspondence analysis showing the relationship between environmental variables, 9 subsamples, and microbial key taxa. Nomenclature of biogeochemical variables is as follows: β-1,4-N-acetylglucosaminidase (NAG), phosphomonoesterase (AP), polyphenol oxidase (POX), ammonium (NH4 +), dissolved organic nitrogen (DON), dissolved organic phosphorous (DOP), and dissolved organic carbon (DOC). Microbial names are indicated as follows: a = Arthrobacter sp., Furcasterigmium furcatum, Phomopsis sp., Clonostachys rosea, Ilyonectria sp., and Trichoderma sp; b = Pseudomonas spp., Fusariella sp., and Mortierellaceae sp; c = Didymellaceae sp., Talaromyces wortmannii, and Parengyodontium album; d = Mortierella turficola, Dothideomycetes sp., Metarhizium carneum, and Wojnowiciella dactylidis; e = Beauveria sp., Gaeumannomyces californicus, Aspergillus inflatus, Metarhizium anisopliae, Nectriaceae sp., Pleosporales sp., and Trichoderma koningii; f = Microbacterium sp., Planococcaceae spp., Setophaeosphaeria hemerocallidis, Parapyrenochaeta acacia, and Aureobasidium pullulans; g = Pseudomonas sp., Planococcaceae sp., and Bacillus sp.; h = Pleosporales sp., and Xanthomonadaceae sp.; I = Planococcaceae sp., and Parengyodontium album; Cla = Cladosporium sp., Cla1 = Clavicipitaceae sp., Dia = Diaporthaceae sp., Dia1 = Diaporthe sp., Pen = Penicillium sp., Ple = Pleosporales sp., Pmar = Planococcaceae sp., Srh1 = Xanthomonadaceae sp., Srh2 = Xanthomonadaceae sp., Tol = Tolypocladium geodes.
Discussion
Soil microbiota, including bacteria and fungi, plays central roles in soil fertility and promotes plant health via complex cross-kingdom interactions. Nonetheless, microbial diversity in soils remains poorly understood at different spatial scales. Herein we report 52 microbial OTUs that represent several edaphic functional guilds at the small-scale. This culture-based approach provides the opportunity for a posteriori studies and the possibility of ex situ preservation of genetic resources in face of MCF imminent threats.
Compared with culture-dependent studies of soil fungal diversity in cloud forests at the large-scale (e.g., 90 samples across coffee plantations and MCF yielding to 415 species in Arias and Heredia-Abarca [2014]; and 20 samples from 4 forest fragments reporting 233 species in Arias and Heredia-Abarca [2020]), our results suggested moderate culturable diversity levels within a 10 × 10 × 10 m-transect. Remarkably, the occurrence fungal OTUs such as Trichoderma koningii and species of the genera Beauveria, Cladosporium, Penicillium and Trichoderma, agree with former reports on these taxa from conserved and fragmented cloud forest sites (Arias & Heredia-Abarca, 2014, 2020). In terms of prokaryotic diversity, the most abundant genera were Pseudomonas and Bacillus (both potentially phosphate solubilizer bacteria) in agreement with former work in the Santuario del Bosque de Niebla, a protected area of MCF in Veracruz State (Reverchon et al., 2019, 2020).
The obtained fungi included ubiquitous soil saprobes, as well as potential pathogens of insects, plants, and fungi. In this sense, the abundant isolation of entomopathogenic fungi such as T. geodes, Beauveria, Metarhizium, and Trichoderma members, agrees with previous reports from remnants of the original cloud forest in Mexico (Arias & Heredia-Abarca, 2014; Zarza et al., 2022) and may indicate strong antagonistic processes in soil communities (Zimmermann, 1993). In addition, these taxa may be implicated as an important component of edaphic nitrogen dynamics, by mobilizing nitrogen from hosts (e.g., insects) to the soil, resulting in increased nitrogen availability (Behie et al., 2012), in accordance to the observed high values of β-1,4-N-acetylglucosaminidase -chitinolytic enzyme involved in C and N-acquiring microbial activities that is highly correlated with fungal biomass (Miller et al., 1998; Parham & Deng, 2000; Sinsabaugh & Findlay, 1995).
Given the importance of prompt disease detection and identification of ethological agents in phytopathology, particularly for endemic species inhabiting fragile ecosystems (such as A. salvinii), as a marginal result we present the first report of Clonostachys rosea as a possible phytopathogen of A. salvinii. This fungus has been identified as a phytopathogen of numerous hosts including faba bean (Afshari & Hemmati, 2017), Gastrodia elata (Lee et al., 2020), soybean (Bienapfl et al., 2012) and the fern Sphaeropteris lepifera (Guu et al., 2010); remarkably causing disease under environmental conditions similar to MCF. In this work, we detected the occurrence of C. rosea in soil samples and on sick fronds of A. salvinii, alerting about a possible emerging disease that should be further monitored.
Overall, we did not detect sharp spatial patterns at the small-scale in the analyzed microbial communities, resembling previous observations on the marked spatial heterogeneity at this scale in soil communities (e.g., Nielsen et al., 2010). Particularly, the CCA results for bacteria depicted no particular influence of the tested biogeochemical factors on bacteria. This highlights the need of understanding how small-scale environmental heterogeneity underlies microbial species richness in MCF. Nonetheless, in accordance with our hypothesis, we observed that some microbial players were strongly associated with particular soil biogeochemical variables. For example, T. geodes (entomopathogen) was associated with DON. This is relevant, as Tolypocladium members are known as key players in denitrification processes (Jirout, 2015). Furthermore, the association between M. turficola -plant growth promoting fungus (Ozimek & Hanaka, 2021)-, M. carneum (entomopathogen), and W. dactylidis -potentially phytopathogenic (Marin-Felix et al., 2019)-, with NH4 + in samples collected near A. salvinii might indicate their contribution to the regulation of edaphic inorganic N in the proximities of this fern.
The copious isolation of antagonic OTUs such as T. geodes, Metarhizium spp., and M. turficola -taxa secreting siderophores, a peptide with potential for biological control of fungi and bacteria (Ozimek & Hanaka, 2021)-, agrees with former metabarcoding data (Velez et al., 2021), and suggests imperative in situ biotic regulatory processes of ecosystem functioning, which should be further confirmed by experimental work. In this sense, research on microbial interactions should shed light into building models to predict the outcome of community alterations and the effects of perturbations (Faust & Raes, 2012).
Soil microbial community in the examined pristine Mexican MCF was dominated by potentially entomopathogenic fungal taxa such as T. geodes, and theoretically phosphate solubilizer bacteria such as Pseudomonas and Bacillus spp. In accordance with our hypothesis, microbial assemblages were associated with soil biogeochemical variables such as DON, DOC, POX and NH4 +. The lack of small-scale community structure patterns coupled to a strong environmental heterogeneity, even at the small-spatial scale, is of vital significance and should be considered for the development and application of in situ conservation strategies. This is the first report of C. rosea as a phytopathogen of A. salvinii, which could pose a threat to the communities of this emblematic plant species. In the future, the possible utilization of the herein isolated native microbial genetic resources ought to be probed to evaluate their response to shifting environmental conditions, as well as for their relationship with plant hosts.

