











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Epilepsy is a central nervous system (CNS) dysfunction syndrome caused by synchronized overdischarge of brain neurons due to a variety of reasons, with recurrent, stereotyped, paroxysmal, and transient characteristics1,2. At present, there are two hypotheses to explain refractory epilepsy resistance, the multidrug-resistant transporter hypothesis in the brain, and the sensitivity reduction of drug targets hypothesis3,4. Therefore, the adenosine system, as a regulator of endogenous antiepileptic effects in the brain, has attracted widespread attention. Adenosine A1 receptor (AA1R) in the adenosine receptor family is widely present in the CNS, especially with high expressions in the cerebral cortex and hippocampus5, exerting brain protective and antiepileptic effects, mainly by binding corresponding G proteins. If exogenous drugs are used to activate AA1R or to increase the concentration of endogenous adenosine, AA1R can protect the brain and combat epilepsy, but the underlying mechanisms are still largely unknown6-8. Epileptic seizures are closely related to neuronal apoptosis. After seizures, CNS neurons are selectively damaged, leading to plastic changes in functional structures such as glial proliferation, mossy fiber budding and synaptic structural remodeling. In addition, these changes may aggravate seizures, eventually causing uncontrollable and recurrent epilepsy9. GABA receptor is a crucial inhibitory neurotransmitter of the CNS, the level of abnormality of which has been associated with many neurological and psychiatric diseases, also with sedative and anxiolytic effects10. Stubbs et al. reported that increasing the expression of GABA receptor, a target of antiepileptic drugs, could control epileptic seizures, but its relationship with AA1R remained elusive11. AA1R can protect against neuronal injury of ischemic cerebrovascular diseases through various molecular biological mechanisms. However, whether AA1R has neuroprotective effects on seizure-induced hippocampal neuronal injury is still unclear. Thereby motivated, we herein assessed the neuroprotective effects of AA1R on hippocampal neuronal injury after lithium chloride-pilocarpine-induced epilepsy.
MATERIALS AND METHODS
Experimental animals
Male SD rats (Sprague Dawley rats grade) weighing 200-250 g were purchased from Beijing, Experimental Animal Breeding Center (China). The animals were kept under the following standard experimental conditions: a light/dark cycle of 12 h/12 h; without noise; feeding in individual cages; temperature of 18-25°C; and relative humidity of 40-70%. The experimental process was reviewed and approved by the ethics committee of our hospital. The rats were given adequate food and water before and after the experiment, and their pain was minimized throughout the experiment.
Animal grouping
A total of 60 male rats were randomly divided into four groups of 15 rats each: a control group, an epilepsy group, an epilepsy + AA1R antagonist (DPCPX) group, and an epilepsy + AA1R agonist (2-CAdo) group. The four groups were observed at three time points, on days 1, 14, and 30, with five rats for each time point.
The epilepsy group was given 125 mg/kg lithium chloride by intraperitoneal injection and 20 mg/kg pilocarpine 20 h later. The Racine staging was employed for categorizing epileptic seizures: Stage 0: without behavioral changes; Stage 1: mouth and facial movement; Stage 2: head nodding; Stage 3: forelimb clonus; Stage 4: rearing with forelimb clonus; and Stage 5: rearing and falling with forelimb clonus12. The epilepsy model was considered successful if Stage 4-5 seizures occurred. The rats were given atropine (1 mg/kg) by intraperitoneal injection 30 min after continuous seizures. 30 min later, they were further injected intraperitoneally with diazepam (10 mg/kg) and 10% chloral hydrate (5 mg/kg) during a convulsion in case of imminent danger. After the seizures were controlled, the rats were subjected to heat preservation and fed in individual cages after waking up, with free access to food and water. For the epilepsy + DPCPX group, DPCPX was injected into the tail vein 1 h before intraperitoneal injection of lithium chloride-pilocarpine and 1 h after convulsions at a dose of 0.3 mg/kg. As to the epilepsy + 2-CAdo group, 2-CAdo was injected into the tail vein 1 h before intraperitoneal injection of lithium chloride-pilocarpine and 1 h after convulsions at a dose of 0.6 mg/kg.
Histopathological procedures
All rats were sacrificed under anesthesia to obtain the brain tissue specimen which was fixed with 10% formalin for 24 h to prepare paraffin sections. The sections were deparaffinized with xylene and rinsed with water after each concentration of ethanol: xylene I, II, 5 min each; absolute ethanol twice, 2 min each; 95% ethanol, 1 min; 90% ethanol, 1 min; 80% ethanol, 1 min; and 70% ethanol, 1 min. Subsequently, the sections were rinsed twice with distilled water, stained by hematoxylin for 5 min, rinsed with tap water, and differentiated in hydrochloric acid-ethanol for 10 s, followed by immersion in tap water for 10 min, in 75% ethanol for 2 min, and in 85% ethanol for 2 min. Then, the sections were subjected to eosin staining for 2 min, conventional dehydration and transparency: 95% ethanol, 5 min; absolute ethanol, 5 min; and xylene I, II, 5 min each. Finally, the sections were mounted with neutral resin and observed under a light microscope.
Detection of neuron apoptosis by TUNEL assay
TUNEL assay kit (Thermo Fisher Scientific, USA) was used for the detection of apoptosis. For the hippocampus tissue section of each sample, five visual fields of 400-fold light microscope were randomly selected. After TUNEL staining, the nuclei of positive apoptotic neurons were brown or tan. The total number of cells and the number of TUNEL-positive cells were calculated, respectively, and averaged. The apoptotic index (AI) is the number of TUNEL-positive cells divided by the total number of cells per visual field.
Western blot
The hippocampus was separated from brain tissue, added RIPA lysis buffer (150 µl/20 mg), homogenized, and centrifuged at 12,000 rpm for 5 min. Then, the supernatant was collected into an EP tube, and the protein concentration was measured with a BCA protein quantification kit (Beyotime Institute of Biotechnology, China). Each sample (50 µg) was subjected to sodium dodecyl-sulfate polyacrylamide gel electrophoresis, and the product was electronically transferred onto a nitrocellulose membrane. Subsequently, the membrane was blocked with 5% skimmed milk at room temperature for 1 h, incubated overnight with primary antibodies at 4°C, washed, and incubated with the corresponding secondary antibodies at 37°C for 1 h. After developing and scanning, relative protein expressions were detected by Quantity One software after correction with internal reference GAPDH.
RESULTS
Effects of AA1R on pathological and morphological changes of neurons in the hippocampal CA3 area after injury
On days 1, 14, and 30, neurons in the hippocampal CA3 area of the normal control group were neatly arranged, and the nuclei were round or oval, with intact structures (Fig. 1a).
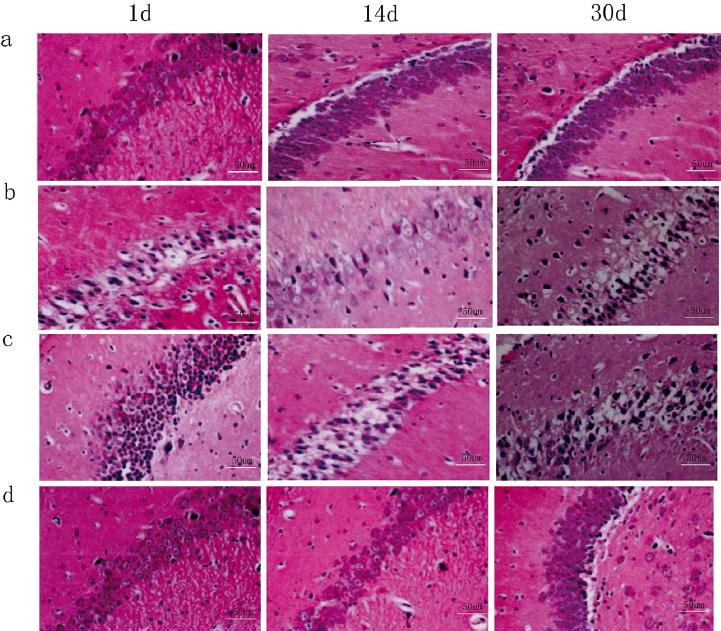
Figure 1 HE staining results of neurons in the hippocampal CA3 area on days 1, 14, and 30. (a) Normal control group, (b) epilepsy group, (c) epilepsy + DPCPX group, (d) epilepsy + 2-CAdo group.
In the epilepsy group, the cellular structure was not neatly arranged, and some neurons were swelling, thick, and incomplete. With prolonged time, the neurons became disorderly arranged, with unclear boundary, nuclear condensation, shrinkage, and cytoplasmic vacuoles. Meanwhile, the neurons lost their normal structure, undergoing evident degeneration (Fig. 1b).
Compared with the epilepsy group at the same time point, the epilepsy + DPCPX group had aggravated distortion, disorganization, edema, cytoplasmic vacuoles, degeneration, and other structural damages (Fig. 1c). In the epilepsy + 2-CAdo group, the cell arrangement was basically regular and orderly, and structural damages, such as edema, cytoplasmic vacuoles, and degeneration, were lessened (Fig. 1d).
Antiapoptotic effects of AA1R on neurons in the hippocampal CA3 area after injury
On days 1, 14, and 30, TUNEL assay of hippocampal CA3 neurons showed that AI values of the normal control group on days 1, 14, and 30 were similar (p > 0.05) (Fig. 2a and Table 1). Compared with the normal control group at the same time point, the epilepsy group underwent evident neuronal apoptosis, with a significantly higher AI (p < 0.05) (Fig. 2b and Table 1).
Table 1 AI values of neurons in the hippocampal CA3 area on days 1, 14, and 30 (%).
| Group | AI (%) | ||
|---|---|---|---|
| Day 1 | Day 14 | Day 30 | |
| Normal control | 7.15±0.17 | 8.07±0.21 | 7.67±0.19 |
| Epilepsy | 19.87±2.18a | 33.41±1.98a | 41.23±1.88a |
| Epilepsy + DPCPX | 29.86±2.09b | 40.78±1.96b | 48.81±1.69b |
| Epilepsy + 2-CAdo | 10.97±1.18c | 22.67±1.76c | 25.49±1.79c |
ap < 0.05, comparison between epilepsy and normal control groups at each time point
bp < 0.05, comparison between epilepsy + DPCPX group and epilepsy group at each time point
cp < 0.05, comparison between epilepsy + 2-CAdo group and epilepsy group at each time point
AI: apoptotic index
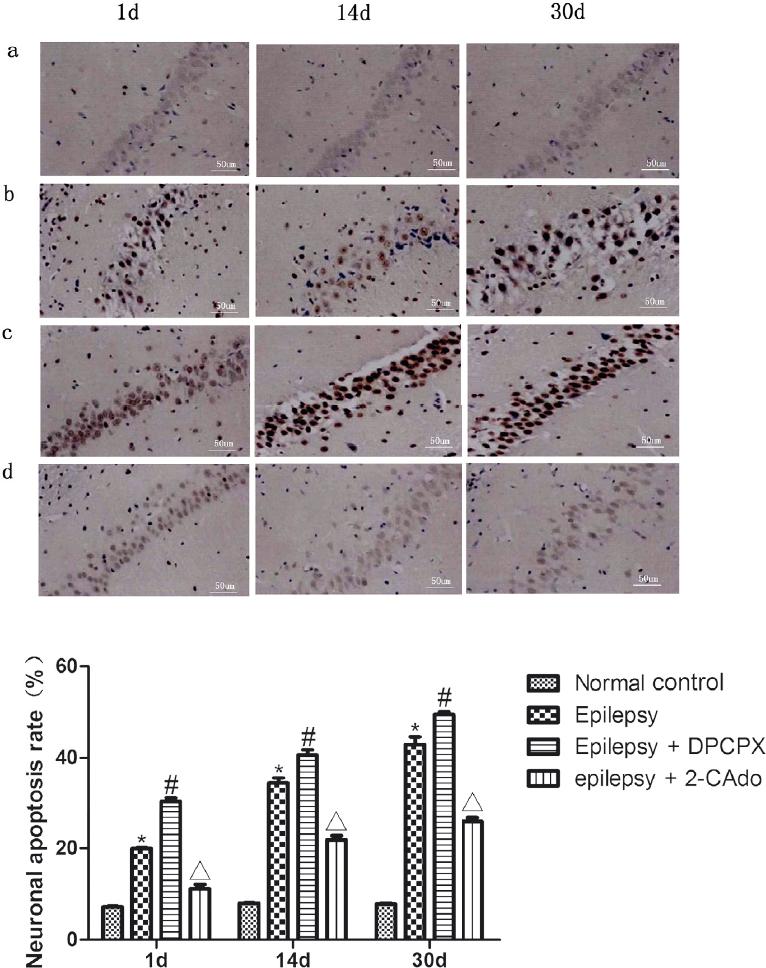
Figure 2 TUNEL staining results of neurons in the hippocampal CA3 area on days 1, 14, and 30. (a) Normal control group, (b) epilepsy group, (c) epilepsy + DPCPX group, (d) epilepsy + 2-CAdo group. *Comparison between normal control and epilepsy groups, p < 0.05; #comparison between epilepsy and epilepsy + DPCPX groups, p < 0.05; Δcomparison between epilepsy and epilepsy + 2-CAdo groups, p < 0.05.
Compared with the epilepsy group, the neuronal apoptosis of the epilepsy + DPCPX group was increased, and AI was also significantly raised (p < 0.05) (Fig. 2c and Table 1). The neuronal apoptosis of the epilepsy + 2-CAdo group was inhibited, and AI was significantly reduced (p < 0.05) (Fig. 2d).
Effects of AA1R on caspase-3 protein expression after injury
Western blot showed that the expressions of caspase-3 in the normal control group on days 1, 14, and 30 were similar (p > 0.05). Compared with the normal control group, the expression levels of the epilepsy group on days 1 and 14 were similar, but levels in day 30 were significantly upregulated (p < 0.05). Compared with the epilepsy group, the expression level of the epilepsy + DPCPX group was similar in day 1, whereas those on days 14 and 30 were significantly upregulated (p < 0.05). Compared with the epilepsy group, the level of the epilepsy + 2-CAdo group was not significantly different in day 1, but those on days 14 and 30 were significantly downregulated (p < 0.05) (Fig. 3).
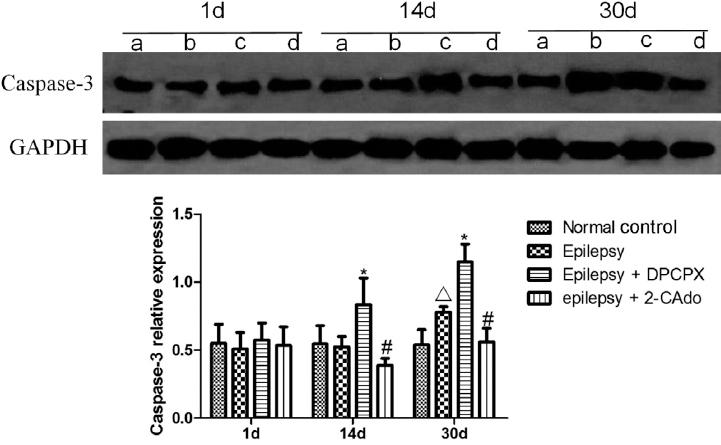
Figure 3 Effects of A1R on caspase-3 protein expression after injury detected by Western blot. (a) Normal control group, (b) epilepsy group, (c) epilepsy + DPCPX group, (d) epilepsy + 2-CAdo group. *Comparison between normal control and epilepsy groups, p < 0.05; #comparison between epilepsy and epilepsy + DPCPX groups, p < 0.05; Δcomparison between epilepsy and epilepsy + 2-CAdo groups, p < 0.05.
Effects of AA1R on GABA receptor expression after injury
The expression levels of GABA receptor in the normal group were similar on days 1, 14, and 30 (p > 0.05). With extended time, the level of the epilepsy group decreased. At the same time point, the level was significantly downregulated compared with that of the normal control group (p < 0.05). Compared with the epilepsy group at the same time point, the GABA expression level of the epilepsy + DPCPX group was significantly downregulated (p < 0.05), whereas that of the epilepsy + 2-CAdo group was significantly upregulated (p < 0.05) (Fig. 4).
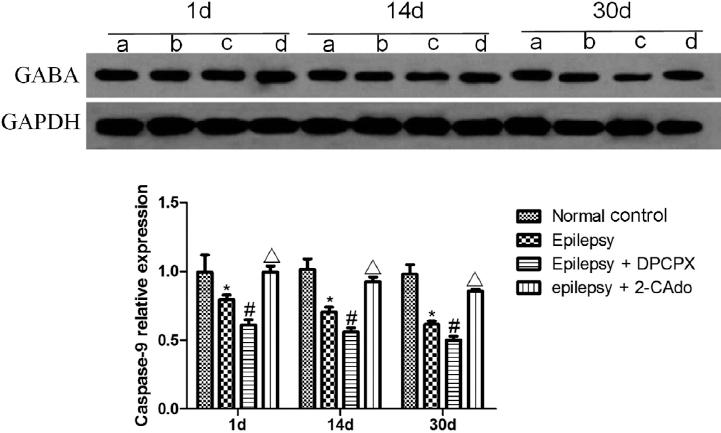
Figure 4 Effects of A1R on GABA receptor expression after injury detected by Western blot. (a) Normal control group, (b) epilepsy group, (c) epilepsy + DPCPX group, (d) epilepsy + 2-CAdo group. *Comparison between normal control and epilepsy groups, p < 0.05; #comparison between epilepsy and epilepsy + DPCPX groups, p < 0.05; Δcomparison between epilepsy and epilepsy + 2-CAdo groups, p < 0.05.
DISCUSSION
The pathogenesis of epilepsy is complex and diverse, which is related to many factors. To date, a variety of epilepsy models have been established, such as those induced by chemical agents and electrical stimulation. The epilepsy model induced by intraperitoneal injection of lithium chloride-pilocarpine has unique pathophysiological processes13. The changes in EEG of rats during seizures are similar to those in humans. The epilepsy model enters the chronic phase 15 days after acute seizures when rats undergo spontaneous seizures in a repeated and intermittent way14-16. In this study, the success rate and mortality rate of the lithium chloride-pilocarpine induction-induced epilepsy model were 82.6% and 10.0%, respectively. In addition, this model has basic pathological changes similar to those of human temporal lobe epilepsy17. It is the most classical chemical-induced animal model for the study of temporal lobe epilepsy, which can specifically damage the hippocampus, causing hippocampal neuronal injury and apoptosis.
Epilepsy is closely related to neuronal apoptosis. Epileptic seizures can induce neuronal apoptosis, which can, in turn, also aggravate the seizures. This study confirmed that neurons were neatly arranged with a clear contour, and the nuclei were round or oval, with intact structures. HE staining of the specimen from the epilepsy group showed that 1 day after seizures, the structure of hippocampal neurons was not neatly arranged, and some cells were swelling, thick, and incomplete. With extended time, the neurons were arranged disorderly on days 14 and 30, with ambiguous contour, unclear boundary, nuclear condensation, shrinkage, cytoplasmic vacuoles, and degeneration. Meanwhile, the gap between cells became wider. Thus, epilepsy indeed caused hippocampal neuronal damage, being consistent with the results of André et al.18 and Hanaya et al.19
In this study, TUNEL assay showed that, compared with the normal control group at the same time point, there was evident neuronal apoptosis in the epilepsy group, which was gradually enhanced on days 1, 14, and 30. Compared with the normal control group, the AI of neurons in the epilepsy group was significantly higher than that of the normal control group (p < 0.05). Thus, seizures induced hippocampal neuronal apoptosis, being in accordance with the study of Patel et al.20 However, the mechanism of neuronal apoptosis after seizures has not yet been fully elucidated, which may be related to the following aspects: (1) seizures lead to neurotransmitter changes in the brain, release considerable excitatory amino acid Glu, which can open channels for Na+, Cl-, and water molecules, causing neuronal swelling followed by neuronal apoptosis; (2) Glu also acts on postsynaptic NMDA and non-NMDA receptors, so Ca2+ enters cells, which can increase intracellular Ca2+ concentration, activate voltage-dependent calcium channels, and ultimately cause neuronal degeneration; (3) Glu increase and calcium ion inflow open the permeable pores of mitochondrial membrane, leading to mitochondrial damage and then neuronal apoptosis. Moreover, opening of the pores aggravates neuronal damage by releasing cytochrome C; and (4) seizures affect the regulation of neuronal apoptosis-related gene expressions and signaling pathways. Using an animal model of lithium chloride-pilocarpine-induced epilepsy, Tan et al. confirmed that P53 gene and caspase-3 were associated with neuronal apoptosis21. Another study also verified that neuronal damage was related to the MAPK signaling pathway after seizures. In short, neuronal damage and apoptosis after seizures are associated with the toxicity of excitatory amino acids, increased ion flow, mitochondrial damage, and the MAPK signaling pathway.
In this study, hippocampal HE staining of the epilepsy + DPCPX group showed that neuronal arrangement was disordered, with cell edema, cytoplasmic vacuoles, degeneration, and other structural damages. TUNEL assay showed that both neuronal apoptosis and AI significantly increased (p < 0.05). In the epilepsy + 2-CAdo group, the cell arrangement was basically regular and orderly, and structural damages such as cellular edema and degeneration were alleviated. Furthermore, neuronal apoptosis and AI significantly decreased (p < 0.05). Hence, AA1R exerted neuroprotective effects on hippocampal neuronal injury after lithium chloride-pilocarpine-induced epilepsy, being closely related to neuronal apoptosis, which could be antagonized.
Apoptosis is a complex protease cascade reaction mediated by the caspase family, especially caspase-3. Herein, compared with the epilepsy group on days 14 and 30, the caspase-3 expression levels of the epilepsy + DPCPX group were significantly upregulated (p < 0.05), but those of the epilepsy + 2-CAdo group were significantly downregulated (p < 0.05). Likewise, Faherty et al. established a rat model of kainic acid-induced epilepsy and found that caspase-3 activity in the hippocampal area was significantly enhanced in day 30, suggesting that neuronal apoptosis was closely associated with caspase-3 expression changes. In contrast, AA1R reduced the expression of caspase-322.
As an inhibitory neurotransmitter of the CNS, GABA receptor works mainly through chloride ion channels. In this study, compared with the epilepsy group at the same time point, the GABA expression level of the epilepsy + DPCPX group was significantly downregulated (p < 0.05), whereas that of the epilepsy + 2-CAdo group was significantly upregulated (p < 0.05). Similarly, Reid et al. reported that AA1R elevated chloride ion permeability by regulating GABA level, thus inducing hyperpolarization of intracellular membrane and producing inhibitory postsynaptic potential to combat epilepsy23.
In summary, AA1R had neuroprotective effects on hippocampal neuronal damage after induction of epilepsy by lithium chloride-pilocarpine, mitigating cell edema, and neuronal apoptosis. Nevertheless, this study was limited because we studied the role of AA1R in the lithium chloride-pilocarpine-induced epilepsy model only, so other models of epilepsy are needed to unravel the underlying mechanism. The findings provide a novel idea for developing new neuroprotective therapies in clinical practice.

