











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
In agriculture, the herbicide atrazine is commonly used [1-5]. The herbicide atrazine requires a long time up to decades to degrade in water. [6] Moreover, atrazine was recently considered as a potential carcinogen by many reports. [7,8] The consumption of contaminated water is a major route of human exposure to the pollutant. [9] It is therefore important to seek new methodologies to eliminate this herbicide from the aquatic environment, currently the residual level for atrazine in groundwater and drinking water has been established. Different methodologies have been reported to remove the contaminant such as bioremediation by bacterial strain FH-1. Recently there are several studies such as phytoremediation, the use of chemical and physicochemical methodologies in order to eliminate this contaminant from the aquatic environment and the soil.10 Adsorption has evolved into one of the most frequently used physical processes for atrazine removal due to its simplicity and easy operation. [11-14] The adsorption is a widely used process that requires low energy and aims to confine diverse compounds using a variety of adsorbent materials. Adsorption of the atrazine has been scarcely studied; some research has been reported. [13, 15, 17]
The design of new adsorbents for the removal of the contaminant has been the theme of theoretical studies. [18] Kumar et al. studied a series of metal complexes containing atrazine, they reported the interactions of atrazine with transition metal ions. Through the FTIR spectral analysis and 3D molecular modeling suggests that the atrazine can coordinate through the nitrogen. [19] Recently Bessac et al. reported the adsorption of atrazine onto two different clay surfaces as pyrophyllite. [20]
Nowadays is emergent the development of new materials that can be used in the adsorption process to remove atrazine from the water. This paper proposes a monolayer triazine-based CTF-0 functionalized with an aluminum cluster (n=2-6) to coordinate with the contaminant. CTF-0 has been scarcely investigated as an adsorber. This material is attractive due to their flexible parameters as controllable C, N, H composition, surface area pore size. [21] The monolayer can be applied in gas adsorption, gas separation and storage. The aluminum clusters show great potential in adsorption due to its highly delocalized valence electrons. [22, 23]
Computational details
All structures in this work were studied by the ONIOM [24] technique, which involved two levels of theory: the high layer, which involves the central cavity, where the aluminum clusters are coordinated together with the atrazine molecule, was treated with combination of the Perdew-Burke-Ernzerhof (PBE0) [25-27]functional and the def2-TZVP basis set, [28] while the rest of the atoms (low layer) were treated with the HF/3-21G* method [29] using the Gaussian 09 program.[30] In order to characterize all optimized structures, we have computed their vibrational modes at the same level of theory. Geometries obtained from ONIOM calculations were used to perform the NBO analysis. The Natural Bond Orbital theory (NBO version 3.1) is implemented in Gaussian 09. [31] The adsorption energy, E ads , is defined as the difference between the energy of the system atrazine with of cluster/ monolayer and the sum of energy of the (cluster/monolayer) and energy of atrazine molecule. The energies of each molecule were calculated with a single point using the Perdew-Burke-Ernzerhof (PBE0) functional and the def2-TZVP basis set. Also, DFT-D3(BJ) Grimme’s dispersion corrections were performed in our calculations. [32] Adsorption energy (E ads) of different systems are calculated with the following equation:
The adsorption energy of the cluster coordinated to CTF-0 is shown in equation 1, where E [AlnCTF-0] is the energy of the cluster-monolayer n=3-4, 6 systems and E CTF-0 is the energy of monolayer, and E Aln corresponds to the energy of each cluster.
The adsorption energy of the atrazine molecule with the systems [AlnCTF-0] n=3-4, 6 is shown in equation 2, where E (atrazine-[AlnCTF-0]) is the total energy of the system of cluster/ monolayer and atrazine. All generated files from Gaussian 09 were analyzed with the Chemcraft program v1.8.
Results and Discussion
Geometry and optimization of CTF-0
The ONIOM technique was used for the optimization of the CTF-0 structure sheet and the systems. This technique has been widely used for the calculation of site structures, transition states, equilibrium geometries energy gaps and adsorption energies for materials with many atoms and generates good results in a short time. [33] The structure was divided into two layers (see Fig. 1), which are treated with different approximations. The layer in an intimate relationship to adsorption of aluminum clusters and atrazine molecule is treated at high-level with the PBE0/def2-TZVP for accuracy, while regions away from the central cavity are treated at lower-level with the HF/3-21G* basis set. The CTF-0 structure is planar with Cs symmetry, the distance between the two N atoms in the middle of the pore is about 4.869 Å. The optimized structure of CTF-0 is depicted in Fig. 1. The C-C bond lengths between pyrazine and tri-s-triazine rings, and C-N in tri-s-triazine rings are 1.476, and 1.326-1.331 Å, respectively, as previously reported. [22,34] Molecular orbitals and their energies are shown in Fig. 2. The chemical potential (η), global hardness (η), and electrophilicity index (η), were calculated to study the reactivity of CTF-0. The layer showed -2.351 for (η), -0.502 for (η) and -5.355 for (η).
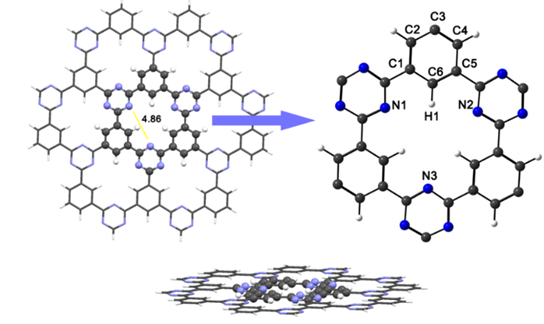
Fig. 1 ONIOM representations for the CTF-0 optimized structure, showing standard model layers. The low-level layer is shown as a wireframe representation, high-level layer is shown as a ball-and-stick representation. Color code for spheres: Monolayer - dark gray (C); blue (N); white (H).

The adsorption of aluminum clusters on CTF-0
We considered clusters with low nuclearity n= 2-6. Our clusters with low energy symmetries agree with the similar aluminum clusters. [35-37] The most stable configurations of Al clusters (Aln) on CTF-0 are discussed, the complexes with Al n = 3 and 5 were positively charged (+1) while Al n = 2, 4 and 6 were neutral. We have intentionally placed all the clusters in the center of the cavity of the CTF-0 surface, the nitrogen atoms which are part of this cavity, have electrons available to interact with aluminum atoms. They were included in the high layer of ONIOM. The lowest energy structures are shown in Figures 3 and 4. In all cluster sizes, ate least two aluminum atoms interact with the carbon and nitrogen atoms of CTF-0. The Al-C bond distances in all the complexes are 2.03-2.92 Å and Al-N 1.89-2.12 Å, see Table 1. It should be noted that the hydrogen atoms located in the central cavity of CTF-0 suffer an out-of-plane distortion causing different C-C-H angles, see Table 1. Such a distortion was attributed to the aluminum clusters.
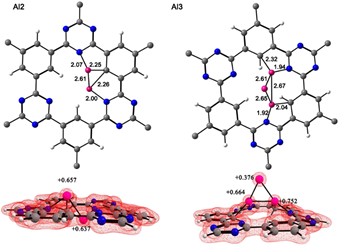
Fig. 3 Top views of aluminum clusters (n= 2-3) on CTF-0 optimized structures and their total charge density. Bond lengths (Å) for aluminum clusters and natural charges for aluminum atoms. Color code for spheres: monolayer - dark gray (C); blue (N); white (H) and pink (Al).
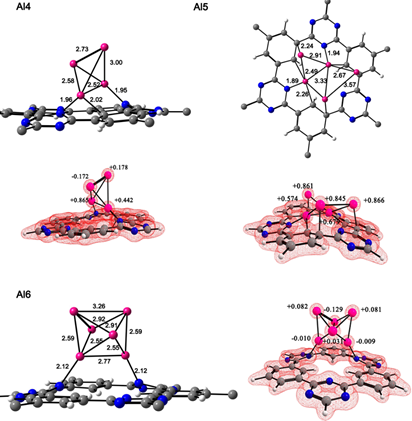
Fig. 4 Top views of aluminum clusters (n= 4-6) on CTF-0 optimized structures and their total charge density. Bond lengths (Å) for aluminum clusters and natural charges for aluminum atoms. Color code for spheres: monolayer - dark gray (C); blue (N); white (H) and pink (Al).
Table 1 Natural charges, Al-N, Al-C bond lengths and C-C-H bond angles of aluminum clusters on CTF-0 calculated with the PBE0/def2-TZVP method.
| Natural charges of Al | Bond lengths (Å) Al-Monolayer | Bond lengths (Å) Al-Al | Bond angles (°) C1-C6-H1 | |
|---|---|---|---|---|
| [Al2CTF-0] | +0.657, +0.637 | Al-N 2.00-2.07 Al-C 2.25-2.26 |
2.61 | 115.1 |
| [Al3CTF-0]+ | +0.376, +0.664, +0.752 | Al-N 1.92-1.94 AL-C 2.04-2.32 |
2.60-2.67 | 133.0 |
| [Al4CTF-0] | -0.172, +0.178, +0.865, +0.442 | Al-N 1.95-1.96 Al-C 2.02-2.92 |
2.44-3.36 | 123.3 |
| [Al5CTF-0]+ | +0.679, +0.861, +0.574, +0.845, +0.866 | Al-N 1.89-1.93 Al-C 2.03-2.75 |
2.49-3.34 | 132.0 |
| [Al6CTF-0] | -0.129, +0.082, +0.081, +0.031, -0.010, -0.009 | Al-N 2.12 | 2.59-3.26 | 121.3 |
In the case of the [Al2CTF-0] complex, Al atoms are coordinated within the cavity, one aluminum atom is coordinated with the N3 oriented at the top of the cavity, while the second Al atom is coordinated with N2 oriented at the bottom of the cavity. The n = 3-6 clusters coordinate at the top of the CTF-0 cavity coordinated with N and C atoms. Coordination of Al clusters with CTF-0 causes a decrease in Al-Al bonds. This behavior is observed more for Al2-Al5 clusters. CTF-0 generates electron density towards the clusters causing the decrease in bonds. The Al-Al distance of the n=2 cluster adsorbed on CTF-0 is less than of the Al2 cluster, showing a linear geometry. The cluster in the [Al3CTF-0]+ system presents a trigonal planar geometry with an Al-Al distance of 2.60-2.70 Å. However, for the [Aln CTF-0] n= 5-6 complex the cluster geometries are modified when absorbed on CTF-0, in the case of the [Al5CTF-0]+ complex, the aluminum cluster shows a better physisorption interaction with the monolayer, forming Al-C bonds. The Al-Al bond lengths, in the [Al6CTF-0] complex, have values of 2.54-2.92 Å, which are in good agreement with those previously reported.38 In n = 4,6 systems some aluminum atoms have negative natural charges. See Fig. 4.
Energies of the highest occupied molecular orbital and lowest unoccupied molecular orbital and HOMO-LUMO energy gaps (Eg) were calculated and presented in Table 2. The HOMO energies for [Al2CTF-0], [Al3CTF-0]+, [Al4CTF-0], [Al5CTF-0]+ and [Al6CTF-0] were -5.25, -7.15, -4.41, -7.73 and -4.57 eV, respectively, while the LUMO energies were between -2.90 and -5.53 eV. On the other hand, for HOMO-LUMO of CTF-0 structures are -7.37 and -2.27 eV. These values indicate that the HOMO energies for these n= 2-4,6 systems increased, as the aluminum cluster interacted with CTF-0. The HOMO-LUMO gap of the monolayer is 4.69 eV, while the HOMO-LUMO gap when aluminum clusters on the monolayer causes a semiconductor character, predominantly for the [Al5CTF-0]+ system with value of 1.09 eV. Table 2 shows adsorption energies for aluminum clusters absorbed on CTF-0. The [AlnCTF-0] n = 2-6 systems have physisorption showing a E ads range of 26.32-91.53 kcal/mol.
Table 2 Adsorption energies (E ads ), frontier molecular orbital energies (E HOMO, E LUMO, eV), HOMO-LUMO energy gaps (Eg, eV), calculated dipole moments (DM, Debye), global hardness (η, eV), chemical potential (μ, eV), and global electrophilicity index (ω, eV) of aluminum clusters on CTF-0 calculated with the PBE0/def2-TZVP method.
| E ads (kcal/mol) | E Homo (eV) | E Lumo (eV) | HOMO-LUMO gaps (eV) | DM (debye) | ( (eV) | ( (eV) | ( (eV) | |
|---|---|---|---|---|---|---|---|---|
| [Al2CTF-0] | 62.79 | -5.25 | -2.90 | 2.33 | 3.620 | 1.18 | -4.07 | 7.06 |
| [Al3CTF-0]+ | 91.53 | -7.15 | -5.54 | 1.62 | 6.417 | 0.81 | -6.34 | 24.90 |
| [Al4CTF-0] | 60.57 | -4.41 | -2.99 | 1.41 | 9.177 | 0.71 | -3.70 | 9.70 |
| [Al5CTF-0]+ | 75.03 | -7.73 | -5.36 | 2.37 | 5.263 | 1.18 | -6.54 | 18.09 |
| [Al6CTF-0] | 90.10 | -4.31 | -3.21 | 1.09 | 7.119 | 0.55 | -3.76 | 12.95 |
Our results show that monolayer is nonpolar, while the [AlnCTF-0] n=2-6 systems are polar with dipole moments of 2.38-9.18 debye (see Table 2). The global reactivity descriptors are provided in Table 2.
The global hardness (η) value in CFT-0 is higher than [AlnCTF-0] n=2-6 systems, suggesting CTF-0 to be less reactive and more stable compared to the systems. The increase in the electrophilicity index (ω) after the formation of the [AlnCTF-0] n=2-6 systems suggest an increased the reactivity. The chemical potential (μ= (EHOMO+ELUMO)/2), global hardness (η= (ELUMO-EHOMO)/2) and global electrophilicity index (ω=μ2/2η) were calculated from the [AlnCTF-0] n=2-6 systems. [39] The chemical potential of [Al4CTF-0], [Al6CTF-0] and [Al2CTF-0] are lower than that of [Al3CTF-0]+ and [Al5CTF-0]+, this predicts lower stability in [AlnCTF-0] n=2,4,6. The descending order of those ( is the following: [Al2CTF-0] > [Al5CTF-0]+ > [Al3CTF-0]+ > [Al4CTF-0] > [Al6CTF-0]. The total charge density is plotted in Figures 3 and 4. These plots confirm that the interaction between CTF-0 and Aln clusters (n = 2-6).
We performed a NBO analysis, only for the internal cavity of CTF-0 and the aluminum clusters. The study of natural bond orbital shows that the π C-C and σ C-N from the monolayer participate as donors, while the aluminum atoms as acceptors see Fig. 5. Interestingly, the NBO analysis shows a back-donation from the aluminum atoms towards the monolayer with their electron donation, similar to the recent reporter by Crimmin, [40] see Table 3. Data show that in all studied interactions, the E(2) energy is higher for [Al5CTF-0]+ due to the formation of seven bonds between Al-C and Al-N showing electron transfer from the monolayer toward the aluminum cluster.
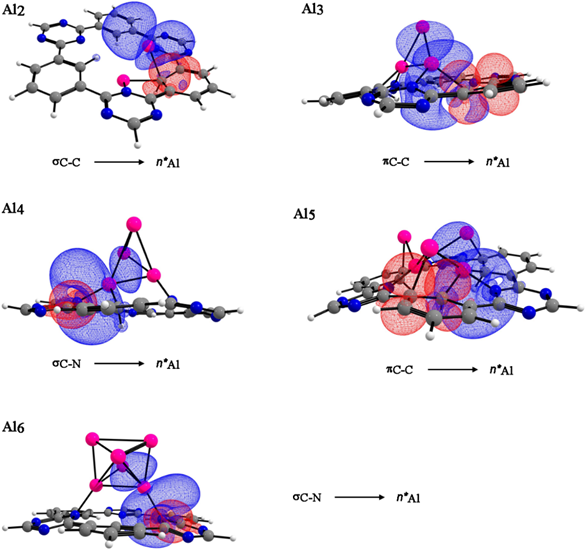
Fig. 5 NBO, donor (red) and acceptor (blue) orbitals responsible for intermolecular interactions of aluminum clusters (n= 2-6) in the optimized CTF-0 structures. Color code for spheres: monolayer - dark gray (C); blue (N); white (H) and pink (Al).
Table 3 Donor and acceptor orbitals for the interactions of aluminum clusters (n= 2-6) on CTF-0 structures.
| Complex | Donor | Acceptor | E(2) (kcal/mol) a | E(j)-E(i) (a.u.) b | F(i, j) (a.u.) c |
|---|---|---|---|---|---|
| [Al2CTF-0] | σC-C | η*Al | 10.02 | 0.74 | 0.078 |
| η*Al | σN-AL | 28.21 | 0.26 | 0.078 | |
| [Al3CTF-0]+ | πC-C | η*Al | 23.39 | 0.06 | 0.056 |
| η*Al | σN-Al | 2.29 | 0.30 | 0.025 | |
| [Al4CTF-0] | σC-N | η*Al | 16.81 | 0.88 | 0.117 |
| η*Al | σN-C | 1.48 | 0.46 | 0.057 | |
| [Al5CTF-0]+ | πC-C | η*Al | 32.42 | 0.12 | 0.066 |
| η*Al | σC-Al | 18.56 | 0.02 | 0.030 | |
| [Al6CTF-0] | σC-N | η*Al | 11.91 | 0.87 | 0.102 |
| η*Al | σC-C | 8.34 | 0.06 | 0.028 |
aE(2) means energy of hyper conjugative interaction (stabilization energy)
bEnergy difference between donor and acceptor i and j BNO orbitals
cF(i, j) is the Fock matrix element between i and j NBO orbitals
The adsorption of atrazine molecule on [AlnCTF-0] n=2-6
In the present study, an atrazine molecule was coordinated through the N3 nitrogen atom (see Fig. 6) to the aluminum clusters supported on the [AlnCTF-0] n=2-6 systems. The geometrical parameters and the adsorption energies of atrazine on the complexes are presented in Table 4. The [AlnCTF-0] n= 3-4,6 systems show coordination with the molecule, observing adsorption energies around 30.67 - 61.08 kcal/mol. The atrazine molecule, upon coordination with the [Al2CTF-0], [Al5CTF-0]+ systems, causes a dissociation of the n=2,5 clusters. These E ads values are higher compared to those previously reported for atrazine adsorbed on pyrophyllite and Ca2+-montmorillonite clay surfaces. [20] The optimized structures of the atrazine-[AlnCTF-0] complexes are shown in Fig. 6. As a result of the geometry optimization, the atrazine- [AlnCTF-0] n= 3-4 complexes, the coordination is vertical between the upper aluminum atom with the N3 of the atrazine molecule with a bond length of 1.96 and 1.99 Å, respectively. In the case of atrazine adsorption on the [Al6CTF-0] complex, its coordination mode is perpendicular with the Al-N bond length, with value of2.00 Å. When an atrazine molecule is adsorbed on [Al3CTF-0]+, the Al-Al (2.44-2.51 Å) bond lengths are shorter than those for the single cluster. The adsorption of atrazine on [Al4CTF-0] and [Al6CTF-0] causes a structural change in the aluminum cluster, showing a stronger interaction of the Al4 cluster with CTF-0. The corresponding distances between the aluminum cluster n = 4 and the CTF-0 monolayer are 1.89-1.98 Å. This system forms three Al-N bonds. The HOMO-LUMO gaps for [AlnCTF-0] n=3,4 (1.62, 1.47 eV) increases to 2.10, 1.54 eV upon interaction with atrazine. On the other hand, the HOMO-LUMO gaps for the [Al3CTF-0]+ system is 1.62 eV and the [Al4CTF-0] system is 1.41 eV. After interaction with the atrazine molecule the HOMO-LUMO gaps increases by 2.10 and 1.54 eV, respectively. The quantum molecular descriptors for adsorption of atrazine on [AlnCTF-0] n=3,4,6 systems are provided in Table 4. The hardness for all systems increases after the adsorption of atrazine. The ionization potential of atrazine-[AlnCTF-0] n=3, 4, 6 systems decrease and the electrophilicity increase in the value of electrophilicity, this indicates the stability for the systems.
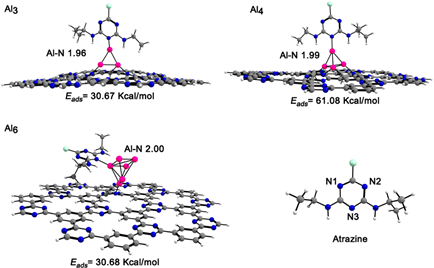
Fig. 6 Side views of the atrazine molecule adsorbed on the [AlnCTF-0] n=3,4, 6 complexes. Al-N bond lengths are given in Å. Color code for spheres: gray (C); pink (Al); white (H); nitrogen (blue) and green (Cl).
Table 4 Adsorption and HOMO-LUMO gaps, and bond lengths of atrazine on complexes, global hardness (η, eV), chemical potential (μ, eV), and global electrophilicity index (ω, eV) calculated with the PBE0/def2-TZVP method.
| Al-atrazine | E ads (kcal/mol) | HOMO-LUMO gaps (eV) | (Å) Al-N atrazine | (Å) Al-Al | η (eV) | μ(eV) | ω (eV) |
|---|---|---|---|---|---|---|---|
| [Al3CTF-0]+ | 30.67 | 2.10 | 1.96 | 2.44-2.52 | 1.06 | -6.05 | 17.25 |
| [Al4CTF-0] | 61.08 | 1.54 | 1.99 | 2.46-2.66 | 0.84 | -3.15 | 5.90 |
| [Al6CTF-0] | 30.68 | 1.03 | 2.00 | 2.48-3.64 | 0.62 | -3.47 | 9.75 |
The NBO analysis of the donor-acceptor interactions are mainly dominated by the nitrogen atoms of the atrazine molecule, which acts as donors and the aluminum atoms as acceptors. On the other hand, the [AlnCTF-0] n=3,4,6 systems exhibit back-donation for aluminum clusters on atrazine molecule, see Fig. 7 and Table 5.
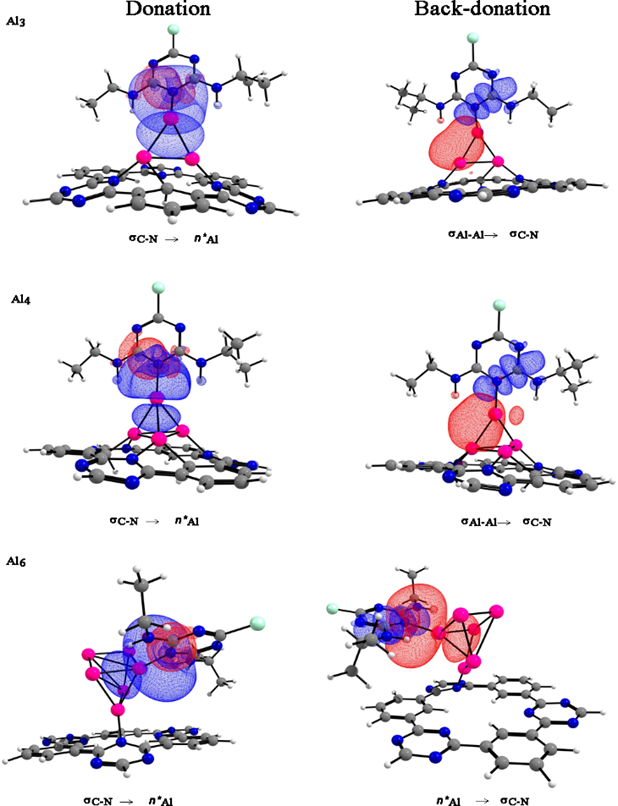
Fig. 7 NBO donor (red) and acceptor (blue) orbitals responsible for intermolecular interactions. Color code for spheres: monolayer - dark gray (C); blue (N); white (H), pink (Al) and green (Cl).
Table 5 Donor and acceptor orbitals for the interactions from atrazine to [AlnCTF-0].
| Molecule | Donor | Acceptor | E(2) (kcal/mol) a | E(j)-E(i) (a.u.) b | F(i, j) (a.u.) c |
|---|---|---|---|---|---|
| Atrazine-[Al3CTF-0]+ | σC-N | η*Al | 12.60 | 0.94 | 0.102 |
| σAl-Al | σ*C-N | 1.07 | 0.74 | 0.027 | |
| Atrazine-[Al4CTF-0] | σC-N | η*Al | 14.60 | 0.88 | 0.112 |
| σAl-Al | σ*C-N | 1.10 | 0.69 | 0.028 | |
| Atrazine-[Al6CTF-0] | σC-N | η*Al | 17.06 | 0.86 | 0.119 |
| η*Al | σ*C-N | 1.05 | 0.48 | 0.044 |
aE(2) means energy of hyper conjugative interaction (stabilization energy)
bEnergy difference between donor and acceptor i and j BNO orbitals
cF(i, j) is the Fock matrix element between i and j NBO orbitals
Contaminate atrazine is found in aqueous media, due to this the adsorption energies of the atrazine molecule in ethanol and water were calculated. All calculations were performed with the PBE0/def2-TZVP method, using the polarizable continuum model (PCM) to simulate the water solvent. [41] For the calculation of the adsorption energy, the same geometry is used for each system, by means of a single point calculation was performed using a PCM model. The adsorption energy of atrazine-[Al6CTF-0] system in water and ethanol is 5 kcal/mol lower than in gas phase, however the system still shows physisorption. On the other hand, adsorption energy values for atrazine-[AlnCTF-0]+ n = 3-4 systems are very similar in the gas phase and in water, see Table 6. When the atrazine molecule coordinates with [AlnCTF-0] n = 3-4 systems, physisorption is observed, because there is no bond breaking in the atrazine molecule and the bond formed is not covalent. [42]
Conclusion
In this work, we have investigated the interaction of aluminum clusters with CTF-0 and the adsorption of atrazine on [AlnCTF-0] n=2-6 complexes using the ONIOM method with the PBE0/def2-TZVP for the high layer. The strongest adsorption was observed for the n=4 aluminum cluster on CTF-0, as an electron transfer from the bond N-C in the cavity toward the aluminum cluster, which means that Al-N and Al-C bonds are strong. When an atrazine molecule is coordinated with the [AlnCTF-0] n=2-6 systems, all geometric parameters show that the atrazine molecule interacts with aluminum atoms to form N-Al coordinative bonds. The physisorption process has been seen in the [AlnCTF-0] n=3-4,6 complexes. The interaction energy values are 30.87-61.08 kcal/mol, and suggest that the systems with n = 3-4,6 could be used as atrazine removers. According to adsorption energy studies, the interaction of atrazine molecule toward [Al4CTF-0] is energetically more favorable than [AlnCTF-0] n=3,6. Finally, NBO calculations show a charge transfer from atrazine to aluminum clusters and a weak back-donation of aluminum atoms to the atrazine molecule. The present study provides new approach and possibilities for the design of new materials for the adsorption of the herbicide contaminant in seawater.

