











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Bacteria form countless diverse and complex communities that reside within each metazoan species, forming what is known as microbiota (Colston & Jackson, 2016; Lee & Mazmanian, 2010). The microbiota is defined as a set of microorganisms and their genomes that inhabit a particular environment, including animals and humans (Bäckhed et al., 2005). Recent advances in molecular biology have provided the opportunity to characterize entire bacterial communities, including those that cannot be isolated in vitro (Bahrndorff et al., 2016). Currently, the 16S rRNA gene is considered an excellent marker for analyzing bacterial communities from any type of sample (Caporaso et al., 2012).
Microbiota studies have provided ecological and evolutionary information showing a strong link with health and disease (Costa et al., 2012). Recently, studies on bacterial microbiota in wild animals have been of great importance since they help to understand the role that these microorganisms play in the health of populations (Santoro et al., 2006). Reptiles carry a great diversity of gram-negative pathogens and are part of their normal microbiota (Ebani et al., 2005). Different epithelia of the body show dissimilar microbial communities. Each bacterial community within the body varies in population structure depending on its anatomical location (Martín et al., 2014). Bacterial communities have been documented on the mucosal surfaces of reptiles, such as the oral, nasal and cloacal cavities, and the intestine (García-De la Peña, Garduño-Niño et al., 2019; García-De la Peña, Rojas-Domínguez et al., 2019; Mackie et al., 2004; Price et al., 2017; Santoro et al., 2006). However, the presence of a blood bacterial microbiota in healthy reptiles has not yet been documented, which, as in domestic animals, would help to better understand the relationship between these bacteria and the health of their host (Peña-Cearra et al., 2021).
The circulatory system is closed, and for a long time, blood was considered a sterile environment in healthy organisms (Drennan, 1942; Proal et al., 2014). Recently, the presence of bacteria in the blood of domestic animals (Mandal et al., 2016; Scarsella et al., 2020; Vientós-Plotts et al., 2017) and healthy humans (Nikkari et al., 2001) has been documented. This perspective of the existence of a “healthy blood microbiota” has sparked much interest in the scientific community (Païssé et al., 2016), with a growing number of studies exploring the idea that the presence of “foreign” microorganisms in vertebrate blood does not necessarily indicate an infection or a disease state (Castillo et al., 2019).
Desert tortoises (genus Gopherus) play an important ecological role, as their burrows provide shelter for various species of animals that inhabit their ecosystems (Witz et al., 1991). In particular, the Texas tortoise (Gopherus berlandieri) is distributed from southeast Texas and northeast Mexico to the states of Coahuila, Nuevo León, San Luis Potosí and Tamaulipas (Bury & Germano, 1994; Judd & Rose, 1983). Its status is considered threatened according to the Official Mexican Norm 059 (Semarnat, 2010), as Least Concern by the International Union for Conservation of Nature (IUCN), and with a high environmental vulnerability score (EVS) according to Terán-Juárez et al. (2016). As like any other animal species, this tortoise is susceptible to bacterial diseases that affect the health of its populations. Because it is necessary to start with a healthy state for reference to diagnose a disease state, in the present study, the composition and abundance of bacteria in the blood of apparently healthy individuals of wild G. berlandieri were determined using next- generation sequencing (16S rRNA gene) in Tamaulipas, Mexico. It is expected that this information might contribute to the development of health, conservation and management programs that consider this microbiological aspect for the survival of this reptile.
Materials and methods
Tamaulipas is located in the northeastern region of Mexico. This state shares borders with Nuevo León, San Luis Potosí, Veracruz, and Texas in the United States of America. Tamaulipas is the sixth largest state in Mexico, with a territorial area of 80,249 km2 (27°40’45” - 22°12’25” N, 97°08’39” - 100°08’42” W) (INEGI, 2017). Due to its geographical location, this region has a high richness of species and temperate and tropical ecosystems (Toledo-Manzur, 1998). The predominant vegetation is the Tamaulipas Thorny Scrub, covering a large part of its surface (Romero, 1999).
Individuals of G. berlandieri were captured manually from August to November 2019 in 3 localities, San Carlos, Casas, and Llera de Canales (Fig. 1). The soil that occurs in these localities is clay-type with a moderately alkaline pH (Zavala et al., 2015). For each tortoise, the following data were recorded: sex (females have a flat plastron, short tail and absence of ventral chin glands; males have a concave plastron, longer tail and presence of chin glands), length and width of the carapace (using a tape measure) and weight (with a hook scale) (Swann et al., 2002). The physical state of each tortoise was determined following the protocols proposed by Mader (2006).
Figure 1 Geographic location of the study sites of Gopherus berlandieri (yellow triangles) in Tamaulipas, Mexico. A = Municipality of San Carlos, B = Municipality of Casas, C = Municipality of Llera de Canales.
With a syringe of 1 ml (25-27 G), a total of 0.5 ml of blood was extracted from each tortoise by puncture in the jugular vein considering the corresponding antiseptic measures (Mader, 2006). Next, 10 drops of blood were deposited into Bashing-bead tubes containing 750 μl of Xpedition™ Zymo Research™ lysing/stabilizing buffer. Each tube was processed in a TerraLyzer cell disruptor (Zymo Research Corporation) for DNA conservation according to the manufacturer. The tubes were placed in a cooler at 4 °C for transportation. When data and blood collection were complete, the tortoises were released at the site of their capture.
Laboratory work. DNA from the blood samples was extracted using the Zymo Research™ DNA Zymobiomics kit. The quantity and quality of DNA per sample was quantified in a ThermoScientific® nanodrop. Amplification of the V3-V4 region of the 16S rRNA gene was developed using the primers suggested by Klindworth et al. (2013): S-D-Bact-0341-b-S-17, 5´-CCTACGGGNGGCWGCAG-3´ and S-D-Bact-0785-a-A-21, 5´-GACTACHVGGGTATCTAATCC-3´, which produced an amplicon of ~460 bp. These sequences were synthesized with the “overhang” adapters of the Illumina protocol (Illumina, 2019a) as follows: 5´TCGTCGGCAGCGTCAGATGTGTATAAGAGACA GCCTACGGGNGGCWGCAG-3´ and 5´-GTCTCGTGG GCTCGGAGATGTGTATAAGAG ACAGGACTACHV GGGTATCTAATCC-3´ (~550 bp amplicon). The Illumina PCR protocol (2017a) was implemented using 12.5 μl of MyTaqTM Ready Mix 1X (Bioline®), 1 μl of each primer (10 µM), 5 μl of DNA (50 ng total) and 5.5 μl of molecular grade H2O. The following cycle was used: 95 °C for 3 minutes; 25 cycles of 95 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 30 seconds; and 72 °C for 5 minutes in a Labnet MultigeneTM Gradient PCR thermocycler. One microliter of the PCR products was placed on a Bioanalyzer DNA 1000 chip to verify the amplicon size (~550 bp). Amplicon purification was performed using 0.8% Agentcourt® AMPure® XP beads. Subsequently, the amplicons were labeled using Nextera (Illumina, 2019b) following this cycle: 95 °C for 3 min; 10 cycles of 95 °C for 30 sec, 55 °C for 30 sec, and 72 °C for 30 sec; and 72 °C for 5 min. Purification was performed again with 1.2% Agencourt® AMPure® XP beads. One microliter of the library was placed on a Bioanalyzer DNA 1,000 chip to verify the amplicon size, expecting a size of ~ 630 bp. Finally, quantification, normalization (equimolarity) and next-generation sequencing (MiSeq Illumina® of 2 × 250 paired-end reads) were performed, following the protocol for 16S metagenomics (Illumina, 2019a).
Bioinformatic analysis. Sequence analysis was performed in an Oracle VM VirtualBox 5.1.14 virtual machine using Quantitative Insights into Microbial Ecology (QIIME) v.1.9.0 bioinformatics software (Caporaso et al., 2010). Sequence processing included quality review using FASTQC and removal of chimeras using USEARCH (Edgar, 2010). Operational taxonomic units (OTUs) were obtained at 97% similarity using the UCLUST method (Edgar, 2010); a random representative sequence was obtained for each OTU, and the taxonomy was assigned using the EzBioCloud database as a reference (Yoon et al., 2017). Afterward, the absolute abundance of OTUs was obtained to construct the rarefaction curve graph to observe the coverage depth; this graph was made in PAST 4.0 (Hammer et al., 2001). Tables of the relative abundance of OTUs were generated at the phylum, class, order, family, genus, and species levels. For the phylum level, a stacked bar graph was created using RStudio; for class to genus levels, a heatmap was constructed to visualize the relative abundance of the most abundant taxa using the Morpheus program (https://software.broadinstitute.org/morpheus/). Finally, a bibliographic search was conducted to review the available literature of all registered genera and species to determine potential pathogens for tortoises, as well as possible zoonotic agents.
Results
The results are shown for 6 G. berlandieri tortoises (1 female and 5 males), tagged as S1 (female from San Carlos), S2 and S3 (males from San Carlos), S4 and S5 (males from Casas), and S6 (male from Llera de Canales). These individuals showed the following morphological means: shell length = 18.5 cm, shell width = 13.93 cm, and weight = 1.2 kg. The mean altitude where tortoises were captured was 274.7 m asl. The mean for the assembled sequences was 28,822.1, the mean for the bacterial sequences was 13,499.3 and for OTUs was 10071.8 (Table 1). An acceptable coverage depth was observed since all rarefaction curves tended to reach an asymptote near 11,000 sequences (Fig. 2).
Table 1 Information on the bacterial sequences obtained from the blood of 6 Gopherus berlandieri tortoises in Tamaulipas, Mexico. ID = Key of each individual, QR = chimeras removed, QS = quality sequences after chimeras removal, BS = bacterial sequences after taxonomic assignment, OTUs = operational taxonomic units.
| ID | Total | Assembled | QR | QS | BS | OTUs |
|---|---|---|---|---|---|---|
| S1 | 172,820 | 29,708 | 2,060 | 27,567 | 11,242 | 7,236 |
| S2 | 110,910 | 27,405 | 2,150 | 25,172 | 12,306 | 8,490 |
| S3 | 134,376 | 36,096 | 326 | 35,665 | 13,747 | 8,007 |
| S4 | 111,236 | 24,399 | 1,396 | 22,951 | 13,248 | 12,076 |
| S5 | 106,302 | 25,237 | 127 | 25,036 | 16,198 | 14,563 |
| S6 | 155,905 | 30,088 | 466 | 29,549 | 14,255 | 10,059 |
| Mean | 131,924.8 | 28,822.1 | 1,087.5 | 27,656.6 | 13,499.3 | 10,071.8 |
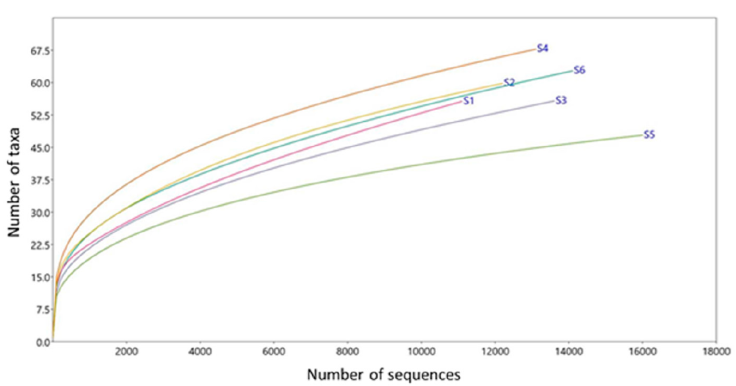
Figure 2 Coverage depth represented as rarefaction curves (number of sequences vs. number of bacterial taxa) obtained from blood samples of 6 Gopherus berlandieri tortoises in Tamaulipas, Mexico.
Nine bacterial phyla were recorded, from which Firmicutes (x̅ = 73.5%), Proteobacteria (x̅ = 13.2%) and Actinobacteria (x̅ = 11.6%) were the most abundant (Fig. 3). A total of 20 classes of bacteria were identified, from which Bacilli (
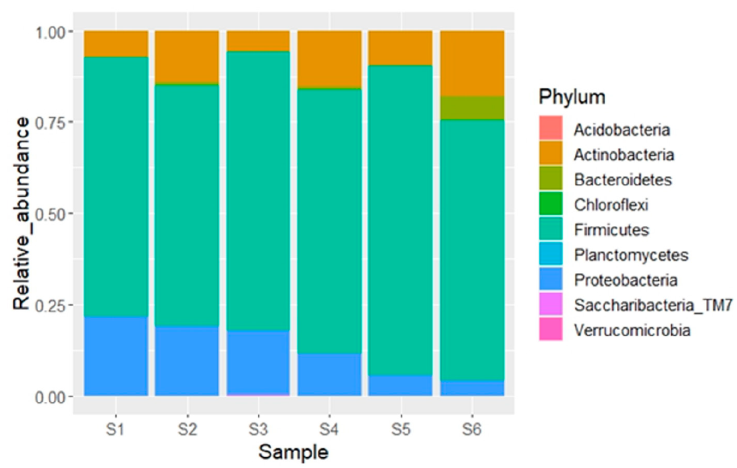
Figure 3 Relative abundance (%) of the bacterial phyla recorded in the blood of 6 individuals of Gopherus berlandieri in Tamaulipas, Mexico.

Figure 4 Heatmap showing the relative abundance of the 40 most abundant bacterial taxa (from class to genera) recorded in the blood of 6 Gopherus berlandieri tortoises in Tamaulipas, Mexico.
Table 2 Bacterial species (only those with a taxonomic name) recorded in the blood of Gopherus berlandieri tortoises in Tamaulipas, Mexico.
| Bacterial species | ||
|---|---|---|
| Acinetobacter colistiniresistens | Bacillus subtilis | Leuconostoc mesenteroides |
| Anaplasma phagocytophilum | Bacillus vietnamensis | Lysobacter ginsengisoli |
| Aquimonas voraii | Bacillus vini | Lysobacter maris |
| Arenimonas aestuarii | Bacteroides timonensis | Lysobacter panaciterrae |
| Arenimonas composti | Beijerinckia derxii | Lysobacter spongiicola |
| Bacillus acidicola | Bilophila wadsworthia | Marinomonas rhizomae |
| Bacillus andreesenii | Caldalkalibacillus thermarum | Moraxella lacunata |
| Bacillus chungangensis | Caldalkalibacillus uzonensis | Nesterenkonia alkaliphila |
| Bacillus cibi | Carboxydocella sporoproducens | Nesterenkonia flava |
| Bacillus crescens | Chiayiivirga flava | Nesterenkonia halotolerans |
| Bacillus dabaoshanensis | Chryseobacterium profundimaris | Nesterenkonia massiliensis |
| Bacillus dakarensis | Clostridium hydrogeniformans | Nesterenkonia rhizosphaerae |
| Bacillus depressus | Cutibacterium acnes | Nocardioides sonneratiae |
| Bacillus endolithicus | Delftia acidovorans | Oceanobacillus oncorhynchi |
| Bacillus halodurans | Effusibacillus pohliae | Ornithinibacillus californiensis |
| Bacillus halosaccharovorans | Finegoldia magna | Paenibacillus barengoltzii |
| Bacillus hemicellulosilyticus | Geobacillus thermantarcticus | Paludicola psychrotolerans |
| Bacillus herbersteinensis | Geobacillus thermodenitrificans | Pontibacillus salicampi |
| Bacillus infantis | Georgenia sediminis | Pseudomonas beteli |
| Bacillus koreensis | Halomonas alkalicola | Rehaibacterium terrae |
| Bacillus licheniformis | Halomonas campaniensis | Salmonella enterica |
| Bacillus luciferensis | Halomonas daqingensis | Sphingopyxis terrae |
| Bacillus massiliosenegalensis | Halomonas desiderata | Staphylococcus capitis |
| Bacillus mesophilum | Halomonas heilongjiangensis | Staphylococcus cohnii |
| Bacillus nanhaiisediminis | Halomonas kenyensis | Staphylococcus equorum |
| Bacillus oceanisediminis | Halomonas nitrilicus | Streptococcus gallolyticus |
| Bacillus okuhidensis | Halomonas stenophila | Thermomonas carbonis |
| Bacillus oleronius | Herbaspirillum huttiense | Undibacterium oligocarboniphilum |
| Bacillus paralicheniformis | Hymenobacter humi | Virgibacillus halodenitrificans |
| Bacillus pseudofirmus | Kocuria polaris | Virgibacillus jeotgali |
| Bacillus shackletonii | Kocuria rosea | Virgibacillus kekensis |
| Bacillus siralis | Kocuria subflava |
Discussion
It has been proposed that the first bacteria that reach the blood of a given vertebrate species come from the mother during pregnancy, when the sibling acquires part of the mother’s bacterial microbiota. Subsequently, other bacteria will reach the blood primarily by physiological translocation, a phenomenon in which live bacteria or their products cross the intestinal barrier, the mucosa of the oral, nasal and urogenital cavities, or the skin of the host into the bloodstream (Blekhman et al., 2015; Emery et al., 2021; Gosiewski et al., 2017; Lloyd-Price et al., 2016; Peña-Cearra et al., 2021). Likewise, some particular bacteria from the microbiota of hematophagous insect vectors (fleas, mosquitoes, and ticks, among others) can enter the circulatory system of vertebrates, sometimes causing diseases in the host (Boulanger et al., 2019; Márquez-Jiménez et al., 2005). In the present study, the phylum Firmicutes was the most abundant in the blood of Gopherus berlandieri. This phylum has been reported to be predominant in the intestine and excrement of G. flavomarginatus (García-De la Peña, Garduño-Niño et al., 2019) and G. polyphemus (Yuan et al., 2015), where they perform sugar fermentation functions (hemicellulose and cellulose) (Sharmin et al., 2013). On the other hand, Firmicutes has also been reported in nasal exudates of G. agassizii, G. berlandieri, G. morafkai and G. polyphemus (Weitzman et al., 2018), as well as in saliva of G. flavomarginatus (García-De la Peña, Rojas-Domínguez et al., 2019). It is possible that bacteria from this phylum enter the bloodstream of G. berlandieri by translocation from the intestinal, nasal, and oral membranes, as proposed by Gosiewski et al. (2017). Likewise, the family Bacillaceae and the genera Caldalkalibacillus, Anaerobacillus, Nesterenkonia, and Bacillus were the most abundant in the blood samples of G. berlandieri. The first 3 genera are common inhabitants of alkaline soils where halophilic vegetation occurs (Bassil & Lloyd, 2019; Borsodi et al., 2015; De Jong, 2020; Edouard et al., 2014). Bacillus is characterized by living in different natural soils (Martin & Travers, 1989; Ohba & Aizawa, 1986), although it has also been reported as an intestinal symbiont in herbivorous vertebrates (Siegel, 2001). Although the bacteria in this soil were not analyzed, it is possible that these 4 genera were part of the substrate where G. berlandieri is active. Since this is a tortoise that digs burrows underground, with a daily contact that this reptile maintains with the soil and, therefore, with its bacteria must be considerably high. It has also been reported that species such as armadillos, badgers, rodents, among other vertebrates and invertebrates can live for short or long periods of time in the burrows of G. berlandieri (Kazmaier et al., 2001). It is likely, that the fecal bacteria of these species remain in the burrows where G. berlandieri could later have contact and acquire them via the oronasal route. Subsequently, it is probable that these soil and fecal bacteria infiltrate the blood through the oral capillaries or even enter through microwounds that the tortoise suffers while feeding on herbs and grasses, which are usually very dry and hard (Forner et al., 2006). Either way, these are environmental bacteria that until now have not been recorded as a possible health risk to tortoises.
Some bacterial taxa recorded in G. berlandieri blood that represent important findings are Coxiella sp., Ehrlichia sp., Anaplasma phagocytophilum, and Salmonella enterica. The first 3 are potential pathogens transmitted by arthropod vectors (ticks, lice, fleas, and mosquitoes) that can infect a wide range of vertebrate species, including reptiles (Jin et al., 2012). Coxiella has been documented as an endosymbiont of several tick species (Daveu et al., 2021). To date, there is only 1 described species (Coxiella burnetii) that is the causal agent of zoonotic Q fever (Široký et al., 2010). Some species of turtles have been found to harbor this bacterium in the oral cavity and cloaca (Emydoidea blandingii, Chrysemys picta, and Terrapene ornata in the United States; Sander et al., 2021). In other chelonians, such as Testudo graeca in Iran, C. burnetii has been recorded in blood, where the tick Hyalomma aegyptium is the vector of this bacterium (Khademi et al., 2023). Likewise, Ehrlichia has been reported in Amblyomma sparsum ticks parasitizing Geochelone pardalis in Zambia, as well as in H. aegyptium ticks parasitizing G. elegans and T. graeca in Jordan (Andoh et al., 2015). Finally, A. phagocytophilum (recorded in the present study in only 1 individual of G. berlandieri) has been previously documented in lizards, snakes, and alligators (Nieto et al., 2009), as well as in 2 wild individuals of G. polyphemus (Raskin et al., 2020). During the manipulation of the G. berlandieri individuals in the present study, no ticks were observed on their bodies. However, it is likely that this tortoise is eventually parasitized by ticks that transmit these bacteria, as in the case of G. agassizii - O. turicata - O. parkeri (Grover & DeFalco, 1995), G. polyphemus - O. turicata (Díaz-Figueroa, 2005), G. polyphemus - A. tuberculatum (Budachetri et al., 2016) and G. flavomarginatus - Ornithodoros turicata (Barraza-Guerrero et al., 2020). Therefore, it is important to determine the species of ticks that parasitize G. berlandieri in the study sites of Tamaulipas, as well as their bacterial microbiota. As a result, the origin of the vector-transmitted bacteria that were recorded in the blood of this tortoise would be established. Additionally, Salmonella enterica is a facultative bacillus that inhabits the intestines of reptiles mostly without causing disease (Lecis et al., 2011). Generally, reptiles acquire these microorganisms orally through contact with contaminated feces, food, water or soil (Pees et al., 2023). This bacterial species has been found in cloacal swabs of G. polyphemus (Lockhart et al., 2008), and in the oral cavity of G. flavomarginatus (García-De la Peña, Rojas-Domínguez et al., 2019); however, this is the first time it has been reported circulating in the bloodstream of G. berlandieri. It can be inferred that S. enterica can possibly enter the blood of this tortoise orally as previously described, and remain there without causing apparent illness.
Finally, it is important to emphasize that the information generated in the present study on the composition and abundance of bacteria in the blood of G. berlandieri joins a list of scientific works that have reported bacteria in the blood of healthy animals, including mice, birds, cats, dogs, and humans (Castillo et al., 2019; Mandal et al., 2016; Sze et al., 2014; Vientós-Plotts et al., 2017). Expectedly, this knowledge will be useful for the conservation and veterinary diagnosis of this chelonian, since it represents the first bacteriological reference in the blood of this tortoise in apparently good health. However, it will be important to periodically evaluate G. berlandieri individuals, not only in Tamaulipas but throughout its geographic distribution, to detect possible signs of microbiological diseases and analyze their blood microbiota during illness. In this way, significant progress could be made regarding wildlife health in northeastern Mexico.

