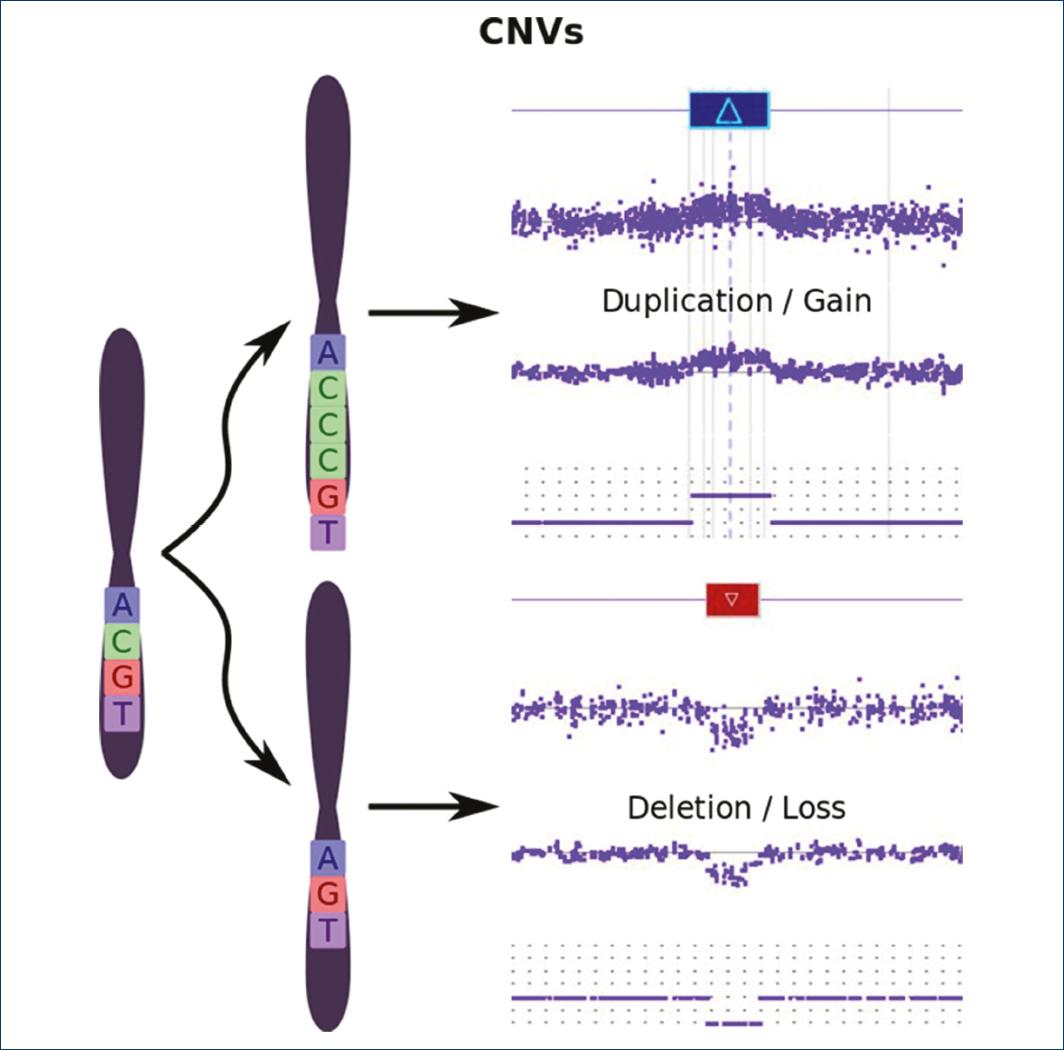
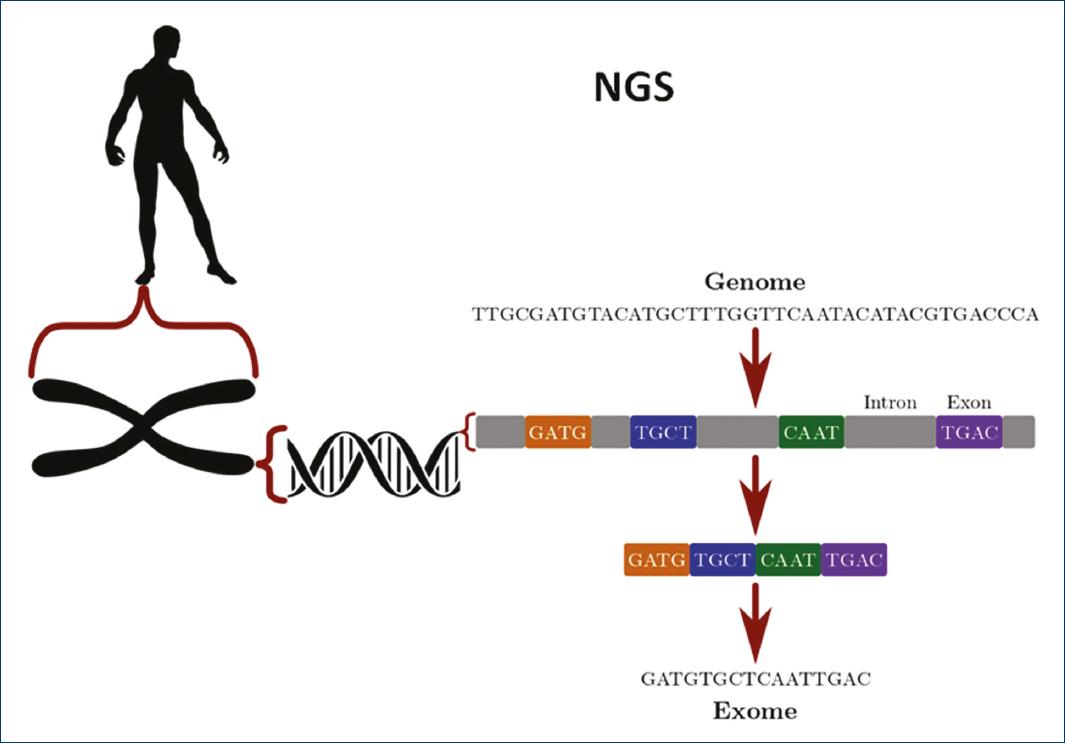
Supplementary material references
1. de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome
sequencing in persons with severe intellectual disability. N Engl J Med.
2012;367:1921-9.
[ Links ]
2. Dimassi S, Labalme A, Ville D, et al. Whole-exome sequencing
improves the diagnosis yield in sporadic infantile spasm syndrome. Clin Genet.
2016;89:198-204.
[ Links ]
3. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et
al. De novo mutations in epileptic encephalopathies. Nature.
2013;501:217-21.
[ Links ]
4. Michaud JL, Lachance M, Hamdan FF, et al. The genetic landscape
of infantile spasms. Hum Mol Genet. 2014;23:4846-58.
[ Links ]
5. Møller RS, Dahl HA, Helbig I. The contribution of next generation
sequencing to epilepsy genetics. Expert Rev Mol Diagn
2015;15:1531-8.
[ Links ]
6. Timal S, Hoischen A, Lehle L, et al. Gene identification in the
congenital disorders of glycosylation Type I by whole-exome sequencing. Hum Mol
Genet. 2012;21:4151-61.
[ Links ]
7. Harvey K, Duguid IC, Alldred MJ, et al. The GDP-GTP exchange
factor collybistin:an essential determinant of neuronal gephyrin clustering. J
Neurosci. 2004;24:5816-26.
[ Links ]
8. Lesca G, Till M, Labalme A, Vallee D, et al. De novo xq11.11
microdeletion including ARHGEF9 in a boy with mental retardation, epilepsy,
macrosomia, and dysmorphic features. Am J Med Genet A.
2011;155A:1706-11.
[ Links ]
9. Lesca G, Depienne C. Epilepsy genetics:the ongoing revolution.
Rev Neurol. 2015;171:539-57.
[ Links ]
10. Shimojima K, Sugawara M, Shichiji M, et al. Loss-of-function
mutation of collybistin is responsible for X-linked mental retardation
associated with epilepsy. J Hum Genet. 2011;56:561-5.
[ Links ]
11. Bruyere H, Lewis S, Wood S, MacLeod PJ, Langlois S. Confirmation
of linkage in X-linked infantile spasms (West syndrome) and refinement of the
disease locus to xp21.3-xp22.1. Clin Genet. 1999;55:173-81.
[ Links ]
12. Claes S, Devriendt K, Lagae L, et al. The X-linked infantile
spasms syndrome (MIM 308350) maps to xp11.4-xpter in two pedigrees. Ann Neurol.
1997;42:360-4.
[ Links ]
13. Feinberg AP, Leahy WR. Infantile spasms:case report of
sex-linked inheritance. Dev Med Child Neurol. 1977;19:524-6.
[ Links ]
14. Fullston T, Brueton L, Willis T, et al. Ohtahara syndrome in a
family with an ARX protein truncation mutation (c.81C>G/p.Y27X). Eur J Hum
Genet. 2010;18:157-62.
[ Links ]
15. Giordano L, Sartori S, Russo S, et al. Familial ohtahara
syndrome due to a novel ARX gene mutation. Am J Med Genet A.
2010;152A:3133-7.
[ Links ]
16. Kato M, Das S, Petras K, et al. Mutations of ARX are associated
with striking pleiotropy and consistent genotype-phenotype correlation. Hum
Mutat. 2004;23:147-59.
[ Links ]
17. Kato M, Saitoh S, Kamei A, et al. A longer polyalanine expansion
mutation in the ARX gene causes early infantile epileptic encephalopathy with
suppression-burst pattern (Ohtahara syndrome). Am J Hum Genet.
2007;81:361-6.
[ Links ]
18. Poeta L, Fusco F, Drongitis D, et al. A regulatory path
associated with X-linked intellectual disability and epilepsy links KDM5C to the
polyalanine expansions in ARX. Am J Hum Genet. 2013;92:114-25.
[ Links ]
19. Proud VK, Levine C, Carpenter NJ. New X-linked syndrome with
seizures, acquired micrencephaly, and agenesis of the corpus callosum. Am J Med
Genet. 1992;43:458-66.
[ Links ]
20. Strømme P, Mangelsdorf ME, Shaw MA, et al. Mutations in the
human ortholog of aristaless cause X-linked mental retardation and epilepsy. Nat
Genet. 2002;30:441-5.
[ Links ]
21. Strømme P, Sundet K, Mørk C, et al. X linked mental retardation
and infantile spasms in a family:new clinical data and linkage to
xp11.4-xp22.11. J Med Genet. 1999;36:374-8.
[ Links ]
22. Turner G, Partington M, Kerr B, Mangelsdorf M, Gecz J. Variable
expression of mental retardation, autism, seizures, and dystonic hand movements
in two families with an identical ARX gene mutation. Am J Med Genet.
2002;112:405-11.
[ Links ]
23. Chan YC, Burgunder JM, Wilder-Smith E, et al.
Electroencephalographic changes and seizures in familial hemiplegic migraine
patients with the CACNA1A gene S218L mutation. J Clin Neurosci.
2008;15:891-4.
[ Links ]
24. Chioza B, Wilkie H, Nashef L, et al. Association between the
alpha(1a) calcium channel gene CACNA1A and idiopathic generalized epilepsy.
Neurology. 2001;56:1245-6.
[ Links ]
25. Holtmann M, Opp J, Tokarzewski M, Korn-Merker E. Human epilepsy,
episodic ataxia Type 2, and migraine. Lancet. 2002;359:170-1.
[ Links ]
26. Jouvenceau A, Eunson LH, Spauschus A, et al. Human epilepsy
associated with dysfunction of the brain P/Q-type calcium channel. Lancet.
2001;358:801-7.
[ Links ]
27. Kors EE, Melberg A, Vanmolkot KR, et al. Childhood epilepsy,
familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation.
Neurology. 2004;63:1136-7.
[ Links ]
28. Chen Y, Lu J, Pan H, et al. Association between genetic
variation of CACNA1H and childhood absence epilepsy. Ann Neurol.
2003;54:239-43.
[ Links ]
29. Heron SE, Khosravani H, Varela D, et al. Extended spectrum of
idiopathic generalized epilepsies associated with CACNA1H functional variants.
Ann Neurol. 2007;62:560-8.
[ Links ]
30. Khosravani H, Altier C, Simms B, et al. Gating effects of
mutations in the cav3.2 T-type calcium channel associated with childhood absence
epilepsy. J Biol Chem. 2004;279:9681-4.
[ Links ]
31. Khosravani H, Bladen C, Parker DB, et al. Effects of cav3.2
channel mutations linked to idiopathic generalized epilepsy. Ann Neurol.
2005;57:745-9.
[ Links ]
32. Vitko I, Chen Y, Arias JM, et al. Functional characterization
and neuronal modeling of the effects of childhood absence epilepsy variants of
CACNA1H, a T-type calcium channel. J Neurosci. 2005;25:4844-55.
[ Links ]
33. Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants
are an important cause of epileptic encephalopathies. Ann Neurol.
2011;70:974-85.
[ Links ]
34. Vergult S, Dheedene A, Meurs A, et al. Genomic aberrations of
the CACNA2D1 gene in three patients with epilepsy and intellectual disability.
Eur J Hum Genet. 2015;23:628-32.
[ Links ]
35. Archer HL, Evans J, Edwards S, et al. CDKL5 mutations cause
infantile spasms, early onset seizures, and severe mental retardation in female
patients. J Med Genet. 2006;43:729-34.
[ Links ]
36. Elia M, Falco M, Ferri R, et al. CDKL5 mutations in boys with
severe encephalopathy and early-onset intractable epilepsy. Neurology.
2008;71:997-9.
[ Links ]
37. Bartnik M, Derwińska K, Gos M, et al. Early-onset seizures due
to mosaic exonic deletions of CDKL5 in a male and two females. Genet Med.
2011;13:447-52.
[ Links ]
38. Erez A, Patel AJ, Wang X, et al. Alu-specific
microhomology-mediated deletions in CDKL5 in females with early-onset seizure
disorder. Neurogenetics. 2009;10:363-9.
[ Links ]
39. Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the
serine/threonine kinase 9 gene causes severe X-linked infantile spasms and
mental retardation. Am J Hum Genet. 2003;72:1401-11.
[ Links ]
40. Nectoux J, Heron D, Tallot M, Chelly J, Bienvenu T. Maternal
origin of a novel C-terminal truncation mutation in CDKL5 causing a severe
atypical form of rett syndrome. Clin Genet. 2006;70:29-33.
[ Links ]
41. Nemos C, Lambert L, Giuliano F, et al. Mutational spectrum of
CDKL5 in early-onset encephalopathies:a study of a large collection of French
patients and review of the literature. Clin Genet.
2009;76:357-71.
[ Links ]
42. Rademacher N, Hambrock M, Fischer U, et al. Identification of a
novel CDKL5 exon and pathogenic mutations in patients with severe mental
retardation, early-onset seizures and rett-like features. Neurogenetics.
2011;12:165-7.
[ Links ]
43. Rosas-Vargas H, Bahi-Buisson N, Philippe C, et al. Impairment of
CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. J Med
Genet. 2008;45:172-8.
[ Links ]
44. Russo S, Marchi M, Cogliati F, et al. Novel mutations in the
CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics.
2009;10:241-50.
[ Links ]
45. Saletti V, Canafoglia L, Cambiaso P, et al. A CDKL5 mutated
child with precocious puberty. Am J Med Genet A.
2009;149A:1046-51.
[ Links ]
46. Scala E, Ariani F, Mari F, et al. CDKL5/STK9 is mutated in rett
syndrome variant with infantile spasms. J Med Genet.
2005;42:103-7.
[ Links ]
47. Tao J, Van Esch H, Hagedorn-Greiwe M, et al. Mutations in the
X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with
severe neurodevelopmental retardation. Am J Hum Genet.
2004;75:1149-54.
[ Links ]
48. Van Esch H, Jansen A, Bauters M, Froyen G, Fryns JP.
Encephalopathy and bilateral cataract in a boy with an interstitial deletion of
xp22 comprising the CDKL5 and NHS genes. Am J Med Genet A.
2007;143:364-9.
[ Links ]
49. Weaving LS, Christodoulou J, Williamson SL, et al. Mutations of
CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and
mental retardation. Am J Hum Genet. 2004;75:1079-93.
[ Links ]
50. Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing
in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1.
Nat Genet. 2013;45:825-30.
[ Links ]
51. Chénier S, Yoon G, Argiropoulos B, et al. CHD2
haploinsufficiency is associated with developmental delay, intellectual
disability, epilepsy and neurobehavioural problems. J Neurodev Disord.
2014;6:9.
[ Links ]
52. Galizia EC, Myers CT, Leu C, et al. CHD2 variants are a risk
factor for photosensitivity in epilepsy. Brain.
2015;138:1198-207.
[ Links ]
53. Liu JC, Ferreira CG, Yusufzai T. Human CHD2 is a chromatin
assembly ATPase regulated by its chromo-and DNA-binding domains. J Biol Chem.
2015;290:25-34.
[ Links ]
54. Lund C, Brodtkorb E, Øye AM, Røsby O, Selmer KK. CHD2 mutations
in lennox-gastaut syndrome. Epilepsy Behav. 2014;33:18-21.
[ Links ]
55. Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations
associated with severe non-syndromic sporadic intellectual disability:an exome
sequencing study. Lancet. 2012;380:1674-82.
[ Links ]
56. Suls A, Jaehn JA, Kecskés A, et al. De novo loss-of-function
mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy
sharing features with dravet syndrome. Am J Hum Genet.
2013;93:967-75.
[ Links ]
57. Thomas RH, Zhang LM, Carvill GL, et al. CHD2 myoclonic
encephalopathy is frequently associated with self-induced seizures. Neurology.
2015;84:951-8.
[ Links ]
58. Trivisano M, Striano P, Sartorelli J, et al. CHD2 mutations are
a rare cause of generalized epilepsy with myoclonic-atonic seizures. Epilepsy
Behav. 2015;51:53-6.
[ Links ]
59. Dibbens LM, Mullen S, Helbig I, et al. Familial and sporadic
15q13.3 microdeletions in idiopathic generalized epilepsy:precedent for
disorders with complex inheritance. Hum Mol Genet.
2009;18:3626-31.
[ Links ]
60. Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions
increase risk of idiopathic generalized epilepsy. Nat Genet.
2009;41:160-2.
[ Links ]
61. Peñagarikano O, Abrahams BS, Herman EI, et al. Absence of
CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core
autism-related deficits. Cell. 2011;147:235-46.
[ Links ]
62. Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive
symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N
Engl J Med. 2006;354:1370-7.
[ Links ]
63. Alakurtti K, Weber E, Rinne R, et al. Loss of lysosomal
association of cystatin B proteins representing progressive myoclonus epilepsy,
EPM1, mutations. Eur J Hum Genet. 2005;13:208-15.
[ Links ]
64. Bespalova IN, Adkins S, Pranzatelli M, Burmeister M. Novel
cystatin B mutation and diagnostic PCR assay in an unverricht-lundborg
progressive myoclonus epilepsy patient. Am J Med Genet.
1997;74:467-71.
[ Links ]
65. de Haan GJ, Halley DJ, Doelman JC, et al. Univerricht-lundborg
(sic) disease:underdiagnosed in the Netherlands. Epilepsia.
2004;45:1061-3.
[ Links ]
66. Di Giaimo R, Riccio M, Santi S, et al. New insights into the
molecular basis of progressive myoclonus epilepsy:a multiprotein complex with
cystatin B. Hum Mol Genet. 2002;11:2941-50.
[ Links ]
67. Lafrenière RG, Rochefort DL, Chrétien N, et al. Unstable
insertion in the 5'flanking region of the cystatin B gene is the most common
mutation in progressive myoclonus epilepsy Type 1, EPM1. Nat Genet.
1997;15:298-302.
[ Links ]
68. Lalioti MD, Mirotsou M, Buresi C, et al. Identification of
mutations in cystatin B, the gene responsible for the unverricht-lundborg type
of progressive myoclonus epilepsy (EPM1). Am J Hum Genet.
1997;60:342-51.
[ Links ]
69. Mazarib A, Xiong L, Neufeld MY, et al. Unverricht-lundborg
disease in a five-generation arab family:instability of dodecamer repeats.
Neurology. 2001;57:1050-4.
[ Links ]
70. Pennacchio LA, Lehesjoki AE, Stone NE, et al. Mutations in the
gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science.
1996;271:1731-4.
[ Links ]
71. Virtaneva K, D'Amato E, Miao J, et al. Unstable minisatellite
expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat Genet.
1997;15:393-6.
[ Links ]
72. Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy
with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol.
2015;77:675-83.
[ Links ]
73. Berkovic SF, Serratosa JM, Phillips HA, et al. Familial partial
epilepsy with variable foci:clinical features and linkage to chromosome 22q12.
Epilepsia. 2004;45:1054-60.
[ Links ]
74. Callenbach PM, van den Maagdenberg AM, Hottenga JJ, et al.
Familial partial epilepsy with variable foci in a dutch family:clinical
characteristics and confirmation of linkage to chromosome 22q. Epilepsia.
2003;44:1298-305.
[ Links ]
75. Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5
cause familial focal epilepsy with variable foci. Nat Genet.
2013;45:546-51.
[ Links ]
76. Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause
autosomal dominant focal epilepsies. Nat Genet. 2013;45:552-5.
[ Links ]
77. Klein KM, O'Brien TJ, Praveen K, et al. Familial focal epilepsy
with variable foci mapped to chromosome 22q12:expansion of the phenotypic
spectrum. Epilepsia. 2012;53:e151-5.
[ Links ]
78. Martin C, Meloche C, Rioux MF, et al. A recurrent mutation in
DEPDC5 predisposes to focal epilepsies in the french-canadian population. Clin
Genet. 2014;86:570-4.
[ Links ]
79. Picard F, Baulac S, Kahane P, et al. Dominant partial
epilepsies. A clinical, electrophysiological and genetic study of 19 European
families. Brain. 2000;123 (Pt 6):1247-62.
[ Links ]
80. Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in
families presenting as autosomal dominant nocturnal frontal lobe epilepsy.
Neurology. 2014;82:2101-6.
[ Links ]
81. Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian
target of rapamycin regulator DEPDC5 cause focal epilepsy with brain
malformations. Ann Neurol. 2014;75:782-7.
[ Links ]
82. Scheffer IE, Phillips HA, O'Brien CE, et al. Familial partial
epilepsy with variable foci:a new partial epilepsy syndrome with suggestion of
linkage to chromosome 2. Ann Neurol. 1998;44:890-9.
[ Links ]
83. Xiong L, Labuda M, Li DS, et al. Mapping of a gene determining
familial partial epilepsy with variable foci to chromosome 22q11-q12. Am J Hum
Genet. 1999;65:1698-710.
[ Links ]
84. Epilepsy Phenome/Genome Project Epi4K Consortium. Copy number
variant analysis from exome data in 349 patients with epileptic encephalopathy.
Ann Neurol. 2015;78:323-8.
[ Links ]
85. Boumil RM, Letts VA, Roberts MC, et al. A missense mutation in a
highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice.
PLoS Genet. 2010;6:e1001046.
[ Links ]
86. Deciphering Developmental Disorders Study. Large-scale discovery
of novel genetic causes of developmental disorders. Nature.
2015;519:223-8.
[ Links ]
87. Dhindsa RS, Bradrick SS, Yao X, et al. Epileptic
encephalopathy-causing mutations in DNM1 impair synaptic vesicle endocytosis.
Neurol Genet. 2015;1:e4.
[ Links ]
88. EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project,
Epi4K Consortium. De novo mutations in synaptic transmission genes including
DNM1 cause epileptic encephalopathies. Am J Hum Genet.
2014;95:360-70.
[ Links ]
89. Perrault I, Hamdan FF, Rio M, et al. Mutations in DOCK7 in
individuals with epileptic encephalopathy and cortical blindness. Am J Hum
Genet. 2014;94:891-7.
[ Links ]
90. Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and
STXBP1:novel genetic causes of dravet syndrome. Neurology.
2014;82:1245-53.
[ Links ]
91. Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an
autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet.
2002;31:184-9.
[ Links ]
92. Ding L, Feng HJ, Macdonald RL, et al. GABA(A) receptor alpha1
subunit mutation A322D associated with autosomal dominant juvenile myoclonic
epilepsy reduces the expression and alters the composition of wild type GABA(A)
receptors. J Biol Chem. 2010;285:26390-405.
[ Links ]
93. Lachance-Touchette P, Brown P, Meloche C, et al. Novel ?1 and ?2
GABAA receptor subunit mutations in families with idiopathic generalized
epilepsy. Eur J Neurosci. 2011;34:237-49.
[ Links ]
94. Maljevic S, Krampfl K, Cobilanschi J, et al. A mutation in the
GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann
Neurol. 2006;59:983-7.
[ Links ]
95. Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and
reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood
absence epilepsy. Am J Hum Genet. 2008;82:1249-61.
[ Links ]
96. Urak L, Feucht M, Fathi N, Hornik K, Fuchs K. A GABRB3 promoter
haplotype associated with childhood absence epilepsy impairs transcriptional
activity. Hum Mol Genet. 2006;15:2533-41.
[ Links ]
97. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2
mutation associated with febrile seizures. Neurology.
2006;67:687-90.
[ Links ]
98. Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic
evidence of GABA(A) receptor dysfunction in epilepsy:a mutation in the
gamma2-subunit gene. Nat Genet. 2001;28:46-8.
[ Links ]
99. Chaumont S, AndréC, Perrais D, Boué-Grabot E, et al.
Agonist-dependent endocytosis of ?-aminobutyric acid Type A (GABAA) receptors
revealed by a ?2(R43Q) epilepsy mutation. J Biol Chem.
2013;288:28254-65.
[ Links ]
100. Chiu C, Reid CA, Tan HO, et al. Developmental impact of a
familial GABAA receptor epilepsy mutation. Ann Neurol.
2008;64:284-93.
[ Links ]
101. Frugier G, Coussen F, Giraud MF, et al. A gamma 2(R43Q)
mutation, linked to epilepsy in humans, alters GABAA receptor assembly and
modifies subunit composition on the cell surface. J Biol Chem.
2007;282:3819-28.
[ Links ]
102. Harkin LA, Bowser DN, Dibbens LM, et al. Truncation of the
GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with
febrile seizures plus. Am J Hum Genet. 2002;70:530-6.
[ Links ]
103. Kananura C, Haug K, Sander T, et al. A splice-site mutation in
GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch
Neurol. 2002;59:1137-41.
[ Links ]
104. Kang JQ, Shen W, Macdonald RL. Why does fever trigger febrile
seizures?GABAA receptor gamma2 subunit mutations associated with idiopathic
generalized epilepsies have temperature-dependent trafficking deficiencies. J
Neurosci. 2006;26:2590-7.
[ Links ]
105. Sancar F, Czajkowski C. A GABAA receptor mutation linked to
human epilepsy (gamma2R43Q) impairs cell surface expression of alphabetagamma
receptors. J Biol Chem. 2004;279:47034-9.
[ Links ]
106. Tan HO, Reid CA, Single FN, et al. Reduced cortical inhibition
in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S
A. 2007;104:17536-41.
[ Links ]
107. Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor
gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet.
2001;28:49-52.
[ Links ]
108. Nakamura K, Kodera H, Akita T, et al. De novo mutations in
GNAO1, encoding a g?o subunit of heterotrimeric G proteins, cause epileptic
encephalopathy. Am J Hum Genet. 2013;93:496-505.
[ Links ]
109. Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause
epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45:1073-6.
[ Links ]
110. Endele S, Rosenberger G, Geider K, et al. Mutations in GRIN2A
and GRIN2B encoding regulatory subunits of NMDA receptors cause variable
neurodevelopmental phenotypes. Nat Genet. 2010;42:1021-6.
[ Links ]
111. Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in
acquired epileptic aphasia and related childhood focal epilepsies and
encephalopathies with speech and language dysfunction. Nat Genet.
2013;45:1061-6.
[ Links ]
112. Lemke JR, Lal D, Reinthaler EM, et al. Mutations in GRIN2A
cause idiopathic focal epilepsy with rolandic spikes. Nat Genet.
2013;45:1067-72.
[ Links ]
113. Scheffer IE, Jones L, Pozzebon M, et al. Autosomal dominant
rolandic epilepsy and speech dyspraxia:a new syndrome with anticipation. Ann
Neurol. 1995;38:633-42.
[ Links ]
114. Nava C, Dalle C, Rastetter A, et al. De novo mutations in HCN1
cause early infantile epileptic encephalopathy. Nat Genet.
2014;46:640-5.
[ Links ]
115. Bassi MT, Balottin U, Panzeri C, et al. Functional analysis of
novel KCNQ2 and KCNQ3 gene variants found in a large pedigree with benign
familial neonatal convulsions (BFNC). Neurogenetics.
2005;6:185-93.
[ Links ]
116. Berkovic SF, Kennerson ML, Howell RA, et al. Phenotypic
expression of benign familial neonatal convulsions linked to chromosome 20. Arch
Neurol. 1994;51:1125-8.
[ Links ]
117. Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel
mutation in neonatal human epilepsy. Science. 1998;279:403-6.
[ Links ]
118. Biervert C, Steinlein OK. Structural and mutational analysis of
KCNQ2, the major gene locus for benign familial neonatal convulsions. Hum Genet.
1999;104:234-40.
[ Links ]
119. Borgatti R, Zucca C, Cavallini A, et al. A novel mutation in
KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation.
Neurology. 2004;63:57-65.
[ Links ]
120. Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions
and epileptic encephalopathy in an italian family with a missense mutation in
the fifth transmembrane region of KCNQ2. Epilepsy Res.
2003;54:21-7.
[ Links ]
121. del Giudice EM, Coppola G, Scuccimarra G, et al. Benign
familial neonatal convulsions (BFNC) resulting from mutation of the KCNQ2
voltage sensor. Eur J Hum Genet. 2000;8:994-7.
[ Links ]
122. Heron SE, Cox K, Grinton BE, et al. Deletions or duplications
in KCNQ2 can cause benign familial neonatal seizures. J Med Genet.
2007;44:791-6.
[ Links ]
123. Saitsu H, Kato M, Koide A, et al. Whole exome sequencing
identifies KCNQ2 mutations in ohtahara syndrome. Ann Neurol.
2012;72:298-300.
[ Links ]
124. Singh NA, Charlier C, Stauffer D, et al. A novel potassium
channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet.
1998;18:25-9.
[ Links ]
125. Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2
encephalopathy:emerging phenotype of a neonatal epileptic encephalopathy. Ann
Neurol. 2012;71:15-25.
[ Links ]
126. Wuttke TV, Jurkat-Rott K, Paulus W, et al. Peripheral nerve
hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology.
2007;69:2045-53.
[ Links ]
127. Yang WP, Levesque PC, Little WA, et al. Functional expression
of two kvLQT1-related potassium channels responsible for an inherited idiopathic
epilepsy. J Biol Chem. 1998;273:19419-23.
[ Links ]
128. Zimprich F, Ronen GM, Stögmann W, et al. Andreas rett and
benign familial neonatal convulsions revisited. Neurology.
2006;67:864-6.
[ Links ]
129. Barcia G, Fleming MR, Deligniere A, et al. De novo
gain-of-function KCNT1 channel mutations cause malignant migrating partial
seizures of infancy. Nat Genet. 2012;44:1255-9.
[ Links ]
130. Derry CP, Heron SE, Phillips F, et al. Severe autosomal
dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders
and intellectual disability. Epilepsia. 2008;49:2125-9.
[ Links ]
131. Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the
sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant
nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188-90.
[ Links ]
132. Ishii A, Shioda M, Okumura A, et al. A recurrent KCNT1 mutation
in two sporadic cases with malignant migrating partial seizures in infancy.
Gene. 2013;531:467-71.
[ Links ]
133. Ohba C, Kato M, Takahashi S, et al. Early onset epileptic
encephalopathy caused by de novo SCN8A mutations. Epilepsia.
2014;55:994-1000.
[ Links ]
134. Vanderver A, Simons C, Schmidt JL, et al. Identification of a
novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy
and severe epilepsy. Pediatr Neurol. 2014;50:112-4.
[ Links ]
135. Chabrol E, Popescu C, Gourfinkel-An I, et al. Two novel
epilepsy-linked mutations leading to a loss of function of LGI1. Arch Neurol.
2007;64:217-22.
[ Links ]
136. Fanciulli M, Santulli L, Errichiello L, et al. LGI1
microdeletion in autosomal dominant lateral temporal epilepsy. Neurology.
2012;78:1299-303.
[ Links ]
137. Fertig E, Lincoln A, Martinuzzi A, Mattson RH, Hisama FM. Novel
LGI1 mutation in a family with autosomal dominant partial epilepsy with auditory
features. Neurology. 2003;60:1687-90.
[ Links ]
138. Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1
cause autosomal-dominant partial epilepsy with auditory features. Nat Genet.
2002;30:335-41.
[ Links ]
139. Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et
al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant
lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119-28.
[ Links ]
140. Nobile C, Michelucci R, Andreazza S, et al. LGI1 mutations in
autosomal dominant and sporadic lateral temporal epilepsy. Hum Mutat.
2009;30:530-6.
[ Links ]
141. Sirerol-Piquer MS, Ayerdi-Izquierdo A, Morante-Redolat JM, et
al. The epilepsy gene LGI1 encodes a secreted glycoprotein that binds to the
cell surface. Hum Mol Genet. 2006;15:3436-45.
[ Links ]
142. Striano P, de Falco A, Diani E, et al. A novel loss-of-function
LGI1 mutation linked to autosomal dominant lateral temporal epilepsy. Arch
Neurol. 2008;65:939-42.
[ Links ]
143. Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile
epileptic encephalopathy caused by mutations in PCDH19 resembles dravet syndrome
but mainly affects females. PLoS Genet. 2009;5:e1000381.
[ Links ]
144. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin
19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet.
2008;40:776-81.
[ Links ]
145. Hynes K, Tarpey P, Dibbens LM, et al. Epilepsy and mental
retardation limited to females with PCDH19 mutations can present de novo or in
single generation families. J Med Genet. 2010;47:211-6.
[ Links ]
146. Kurian MA, Meyer E, Vassallo G, et al. Phospholipase C beta 1
deficiency is associated with early-onset epileptic encephalopathy. Brain.
2010;133:2964-70.
[ Links ]
147. Poduri A, Chopra SS, Neilan EG, et al. Homozygous PLCB1
deletion associated with malignant migrating partial seizures in infancy.
Epilepsia. 2012;53:e146-50.
[ Links ]
148. Shen J, Gilmore EC, Marshall CA, et al. Mutations in PNKP cause
microcephaly, seizures and defects in DNA repair. Nat Genet.
2010;42:245-9.
[ Links ]
149. Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies
truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat
Genet. 2011;43:1252-5.
[ Links ]
150. Heron SE, Grinton BE, Kivity S, et al. PRRT2 mutations cause
benign familial infantile epilepsy and infantile convulsions with
choreoathetosis syndrome. Am J Hum Genet. 2012;90:152-60.
[ Links ]
151. Lee HY, Huang Y, Bruneau N, et al. Mutations in the gene PRRT2
cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep.
2012;1:2-12.
[ Links ]
152. Méneret A, Grabli D, Depienne C, et al. PRRT2 mutations:a major
cause of paroxysmal kinesigenic dyskinesia in the European population.
Neurology. 2012;79:170-4.
[ Links ]
153. Ono S, Yoshiura K, Kinoshita A, et al. Mutations in PRRT2
responsible for paroxysmal kinesigenic dyskinesias also cause benign familial
infantile convulsions. J Hum Genet. 2012;57:338-41.
[ Links ]
154. Pelzer N, de Vries B, Kamphorst JT, et al. PRRT2 and hemiplegic
migraine:a complex association. Neurology. 2014;83:288-90.
[ Links ]
155. Schubert J, Paravidino R, Becker F, et al. PRRT2 mutations are
the major cause of benign familial infantile seizures. Hum Mutat.
2012;33:1439-43.
[ Links ]
156. Striano P, Lispi ML, Gennaro E, et al. Linkage analysis and
disease models in benign familial infantile seizures:a study of 16 families.
Epilepsia. 2006;47:1029-34.
[ Links ]
157. Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang JG, et al.
Identification of PRRT2 as the causative gene of paroxysmal kinesigenic
dyskinesias. Brain. 2011;134:3493-501.
[ Links ]
158. Weber YG, Berger A, Bebek N, et al. Benign familial infantile
convulsions:linkage to chromosome 16p12-q12 in 14 families. Epilepsia.
2004;45:601-9.
[ Links ]
159. Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalized
epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology.
2001;57:2265-72.
[ Links ]
160. Baulac S, Gourfinkel-An I, Picard F, et al. A second locus for
familial generalized epilepsy with febrile seizures plus maps to chromosome
2q21-q33. Am J Hum Genet. 1999;65:1078-85.
[ Links ]
161. Buoni S, Orrico A, Galli L, et al. SCN1A (2528delG) novel
truncating mutation with benign outcome of severe myoclonic epilepsy of infancy.
Neurology. 2006;66:606-7.
[ Links ]
162. Rojo DC, Hamiwka L, McMahon JM, et al. De novo SCN1A mutations
in migrating partial seizures of infancy. Neurology.
2011;77:380-3.
[ Links ]
163. Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in
the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J
Hum Genet. 2001;68:1327-32.
[ Links ]
164. Dichgans M, Freilinger T, Eckstein G, et al. Mutation in the
neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine.
Lancet. 2005;366:371-7.
[ Links ]
165. Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A,
encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet.
2000;24:343-5.
[ Links ]
166. Freilich ER, Jones JM, Gaillard WD, et al. Novel SCN1A mutation
in a proband with malignant migrating partial seizures of infancy. Arch Neurol.
2011;68:665-71.
[ Links ]
167. Mantegazza M, Gambardella A, Rusconi R, et al. Identification
of an nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with
familial simple febrile seizures. Proc Natl Acad Sci U S A.
2005;102:18177-82.
[ Links ]
168. McArdle EJ, Kunic JD, George AL Jr. Novel SCN1A frameshift
mutation with absence of truncated nav1.1 protein in severe myoclonic epilepsy
of infancy. Am J Med Genet A. 2008;146A:2421-3.
[ Links ]
169. Moulard B, Buresi C, Malafosse A. Study of the voltage-gated
sodium channel beta 1 subunit gene (SCN1B) in the benign familial infantile
convulsions syndrome (BFIC). Hum Mutat. 2000;16:139-42.
[ Links ]
170. Mulley JC, Scheffer IE, Petrou S, et al. SCN1A mutations and
epilepsy. Hum Mutat. 2005;25:535-42.
[ Links ]
171. Ohmori I, Ouchida M, Ohtsuka Y, Oka E, Shimizu K. Significant
correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy.
Biochem Biophys Res Commun. 2002;295:17-23.
[ Links ]
172. Orrico A, Galli L, Grosso S, et al. Mutational analysis of the
SCN1A, SCN1B and GABRG2 genes in 150 italian patients with idiopathic childhood
epilepsies. Clin Genet. 2009;75:579-81.
[ Links ]
173. Petrovski S, Scheffer IE, Sisodiya SM, et al. Lack of
replication of association between scn1a SNP and febrile seizures. Neurology.
2009;73:1928-30.
[ Links ]
174. Schlachter K, Gruber-Sedlmayr U, Stogmann E, et al. A splice
site variant in the sodium channel gene SCN1A confers risk of febrile seizures.
Neurology. 2009;72:974-8.
[ Links ]
175. Vahedi K, Depienne C, Le Fort D, et al. Elicited repetitive
daily blindness:a new phenotype associated with hemiplegic migraine and SCN1A
mutations. Neurology. 2009;72:1178-83.
[ Links ]
176. Zucca C, Redaelli F, Epifanio R, et al. Cryptogenic epileptic
syndromes related to SCN1A:twelve novel mutations identified. Arch Neurol.
2008;65:489-94.
[ Links ]
177. Berkovic SF, Heron SE, Giordano L, et al. Benign familial
neonatal-infantile seizures:characterization of a new sodium channelopathy. Ann
Neurol. 2004;55:550-7.
[ Links ]
178. Heron SE, Crossland KM, Andermann E, et al. Sodium-channel
defects in benign familial neonatal-infantile seizures. Lancet.
2002;360:851-2.
[ Links ]
179. Kamiya K, Kaneda M, Sugawara T, et al. A nonsense mutation of
the sodium channel gene SCN2A in a patient with intractable epilepsy and mental
decline. J Neurosci. 2004;24:2690-8.
[ Links ]
180. Liao Y, Anttonen AK, Liukkonen E, et al. SCN2A mutation
associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and
pain. Neurology. 2010;75:1454-8.
[ Links ]
181. Malacarne M, Gennaro E, Madia F, et al. Benign familial
infantile convulsions:mapping of a novel locus on chromosome 2q24 and evidence
for genetic heterogeneity. Am J Hum Genet. 2001;68:1521-6.
[ Links ]
182. Ogiwara I, Ito K, Sawaishi Y, et al. De novo mutations of
voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies.
Neurology. 2009;73:1046-53.
[ Links ]
183. Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense
mutation of the na+channel alpha II subunit gene na(v)1.2 in a patient with
febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S
A. 2001;98:6384-9.
[ Links ]
184. Blanchard MG, Willemsen MH, Walker JB, et al. De novo
gain-of-function and loss-of-function mutations of SCN8A in patients with
intellectual disabilities and epilepsy. J Med Genet.
2015;52:330-7.
[ Links ]
185. de Kovel CG, Meisler MH, Brilstra EH, et al. Characterization
of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy
Res. 2014;108:1511-8.
[ Links ]
186. Veeramah KR, O'Brien JE, Meisler MH, et al. De novo pathogenic
SCN8A mutation identified by whole-genome sequencing of a family quartet
affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet.
2012;90:502-10.
[ Links ]
187. Arsov T, Mullen SA, Rogers S, et al. Glucose transporter 1
deficiency in the idiopathic generalized epilepsies. Ann Neurol.
2012;72:807-15.
[ Links ]
188. Striano P, Weber YG, Toliat MR, et al. GLUT1 muations are a
rare cause of familial idiopathic generalized epilepsy. Neurology.
2012;78:557-62.
[ Links ]
189. Suls A, Mullen SA, Weber YG, et al. Early-onset absence
epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol.
2009;66:415-9.
[ Links ]
190. Molinari F, Raas-Rothschild A, Rio M, et al. Impaired
mitochondrial glutamate transport in autosomal recessive neonatal myoclonic
epilepsy. Am J Hum Genet. 2005;76:334-9.
[ Links ]
191. Poduri A, Heinzen EL, Chitsazzadeh V, et al. SLC25A22 is a
novel gene for migrating partial seizures in infancy. Ann Neurol.
2013;74:873-82.
[ Links ]
192. Hamdan FF, Saitsu H, Nishiyama K, et al. Identification of a
novel in-frame de novo mutation in SPTAN1 in intellectual disability and
pontocerebellar atrophy. Eur J Hum Genet. 2012;20:796-800.
[ Links ]
193. Nonoda Y, Saito Y, Nagai S, et al. Progressive diffuse brain
atrophy in west syndrome with marked hypomyelination due to SPTAN1 gene
mutation. Brain Dev. 2013;35:280-3.
[ Links ]
194. Saitsu H, Tohyama J, Kumada T, et al. Dominant-negative
mutations in alpha-II spectrin cause west syndrome with severe cerebral
hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet.
2010;86:881-91.
[ Links ]
195. Hamdan FF, Piton A, Gauthier J, et al. De novo STXBP1 mutations
in mental retardation and nonsyndromic epilepsy. Ann Neurol.
2009;65:748-53.
[ Links ]
196. Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the
gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy.
Nat Genet. 2008;40:782-8.
[ Links ]
197. Edvardson S, Baumann AM, Mühlenhoff M, et al. West syndrome
caused by ST3Gal-III deficiency. Epilepsia. 2013;54:e24-7.
[ Links ]
198. Berryer MH, Hamdan FF, Klitten LL, et al. Mutations in SYNGAP1
cause intellectual disability, autism, and a specific form of epilepsy by
inducing haploinsufficiency. Hum Mutat. 2013;34:385-94.
[ Links ]
199. Mignot C, von Stülpnagel C, Nava C, et al. Genetic and
neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and
epilepsy. J Med Genet. 2016;53:511-22.
[ Links ]
200. Basel-Vanagaite L, Hershkovitz T, Heyman E, et al. Biallelic
SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic
corpus callosum. Am J Hum Genet. 2013;93:524-9.
[ Links ]
201. Corbett MA, Bahlo M, Jolly L, et al. A focal epilepsy and
intellectual disability syndrome is due to a mutation in TBC1D24. Am J Hum
Genet. 2010;87:371-5.
[ Links ]
202. Duru N, Iseri SA, Selçuk N, Tolun A. Early-onset progressive
myoclonic epilepsy with dystonia mapping to 16pter-p13.3. J Neurogenet.
2010;24:207-15.
[ Links ]
203. Falace A, Filipello F, La Padula V, et al. TBC1D24, an
ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy.
Am J Hum Genet. 2010;87:365-70.
[ Links ]
204. Guven A, Tolun A. TBC1D24 truncating mutation resulting in
severe neurodegeneration. J Med Genet. 2013;50:199-202.
[ Links ]
205. Milh M, Falace A, Villeneuve N, et al. Novel compound
heterozygous mutations in TBC1D24 cause familial malignant migrating partial
seizures of infancy. Hum Mutat. 2013;34:869-72.
[ Links ]
206. Zara F, Gennaro E, Stabile M, et al. Mapping of a locus for a
familial autosomal recessive idiopathic myoclonic epilepsy of infancy to
chromosome 16p13. Am J Hum Genet. 2000;66:1552-7.
[ Links ]












 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink