











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Smoking is the leading cause of preventable death worldwide and is considered a public health problem. The World Health Organization reports that smoking kills more than 5 million people each year. Moreover, 11% of deaths caused by ischemic cardiomyopathy and > 70% caused by lung cancer and chronic obstructive pulmonary disease (COPD) have smoking in common1.
COPD is one of the ten main causes of morbidity and mortality in adult populations worldwide, with a prevalence of 9% in Europe, > 11% in Latin America, 8.6% in Japan, and 6% in the United States2-4. In Mexico, it is also one of the top ten causes of morbidity and mortality5,6. A review study found that 54-77% of patients with COPD of moderate severity were smokers, while 38-51% of those with very severe stages of the disease also smoked7. Nicotine is among the substances found in cigarettes and produces addiction by first affecting the central nervous system. However, after chronic smoking, the smoker can present an inflammatory syndrome, which slowly progresses, causing multisystem damage8. It has been reported that there are different addiction levels. Some smokers smoke less throughout their lifetime and hardly show symptoms of addiction; they are known as chippers. On the other hand, there are heavy smokers who show high nicotine addiction. In general, the latter are less successful in quitting smoking9.
Globally, one billion people will die from tobacco-related illnesses this century if trends in tobacco use remain unchecked. In lowmiddle-income countries such as Mexico, the prevalence of tobacco smoking has not changed10. The classic model of addiction suggests that cigarette consumption increases to a level where regular nicotine administration helps smokers avoid withdrawal symptoms11. Mexican smokers are more likely to be non-daily smokers and to consume a lower number of cigarettes per day (CPD) compared to smokers from the majority of ethnic groups in other countries. Little is known about their quit behaviors12. Mexico was an early signer of the framework convention on tobacco control and is a world leader in public health approaches to tobacco control13. At the national level, the National Tobacco Control Agency (Oficina Nacional para el Control del Tabaco) has instituted health warnings on cigarette packages, pursued a vigorous prevention and cessation media campaign, and produced detailed economic evaluations of the costs of the tobacco epidemic14. However, despite all these efforts, the smoking prevalence in Mexico has remained unchanged15. According to the Global Burden of Disease 2015, while the smoking prevalence declined between 1990 and 2005, these gains were not maintained. In 2010, we began to see an increase in the prevalence of daily smokers, particularly among those between 25 and 34 years of age and who are female13. Consequently, over 14.3 million Mexican adults (16.4%) currently smoke, and it is expected that 4 million will die from tobacco-related diseases in the next decade if the smoking prevalence remains stable. As described in the most-recent Global Adult Tobacco Survey in Mexico, the static smoking prevalence is in part attributable to the fact that fewer than 10% of Mexican smokers take advantage of evidence-based approaches to smoking cessation15. The potential for reducing the projected morbidity and mortality associated with smoking depends greatly on reaching and treating current smokers.
The combination of counseling and pharmacotherapy has been shown to be one of the most effective and feasible interventions for smoking cessation16. Primary care systems in Mexico follow established guidelines for the identification and treatment of smokers. Although health-care providers are usually encouraged to address smoking with their patients at every visit, most providers fail to initiate cessation treatment, even when cessation resources are available, as they are in Mexico. The lack of time during routine patient care, inadequate training of personnel, and competing patient demands are other factors related to the limited implementation of these cessation guidelines17.
In Mexico, tobacco use is causally related to up to 5% of the annual reported mortality18. For the Mexican Social Security Institute (Instituto Mexicano del Seguro Social), smoking generates an annual cost of up to 7082 million pesos (376 million US dollars) simply to address three of the related diseases (lung cancer, chronic obstructive lung disease, and brain-cardiovascular diseases), which represents 4.3% of the total health-care cost of this institution5. To continue in the path to decrease the smoking prevalence in Mexico, it is essential to expand scientific efforts to understand nicotine dependence beyond what we currently know.
NICOTINE ADDICTION AND COPD ARE COMPLEX AND MULTIFACTORIAL DISEASES
The reinforcement effect of nicotine is the main reason for its consumption. However, between 10% and 20% of smokers never show addiction. In addition to the environmental and sociocultural context, genetic variants associated with a higher risk of cigarette smoking have been described mainly in Caucasian and African American populations. Studies in twins have demonstrated that the genetic component for cigarette smoking accounts for up to 35%19.
In some cases, nicotine addiction causes an increase in cigarette smoking. Damage to lung function occurs in approximately 40% of smokers, which could result in the development and progression of COPD20. Studies in twins report that 60% of the individual susceptibility to develop COPD depends on genetic factors21. On the other hand, 18.6% of COPD patients have at least one family member with the same pathology and have a higher number of exacerbations and a poorer quality of life22.
Nicotine addiction and COPD are complex and multifactorial diseases, where the genetic component contributes to their development (Fig. 1). To identify genetic variants associated with these types of diseases, there are different genetic strategies, including cohort analysis, twin or family studies, and case/control group analysis.
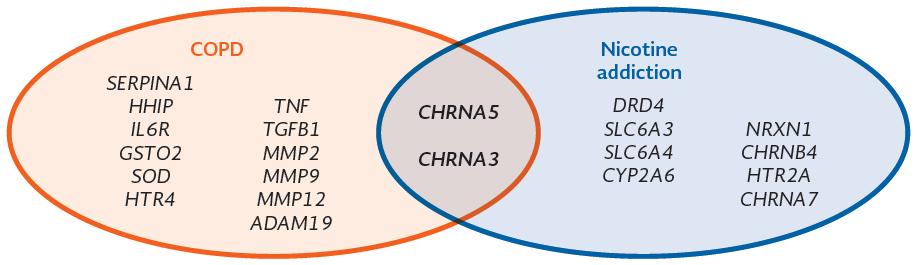
Figure 1 Main genes associated with chronic obstructive pulmonary disease and nicotine addiction, as well as those that have been associated in both pathologies.
Candidate gene studies are one of the older strategies used in genetic association studies. This method evaluates genes that encode proteins involved in biological pathways known in a particular illness. In the case of nicotine addiction, genes encoding for brain neurotransmitter receptors and involved in nicotine metabolism have been evaluated.
With new technological advances in genomics, other methods have been developed to accelerate the identification of genes associated with complex diseases, such as genome-wide association studies (GWASs). GWAS analyze hundreds of thousands to millions of single-nucleotide polymorphisms (SNPs), a strategy that provides a unique opportunity for exploring the genome without any hypothesis23.
POLYMORPHISMS ASSOCIATED WITH NICOTINE ADDICTION IDENTIFIED BY THE CANDIDATE FUNCTIONAL GENE STRATEGY
In the studies of candidate genes, polymorphisms associated with nicotine addiction have been identified in gene encoding proteins involved in the reward and pleasure mechanisms in the central nervous system. The dopaminergic pathway was the first one found among the pathways explored.
The dopamine D4 receptor, specifically a variable number tandem repeats (VNTR) polymorphism, is found among the genes with higher evidence of association with the nicotine addiction process. The biological importance translates to alterations in the length of the receptor. The 48-base pair (bp) VNTR in exon 3 encodes for the third intracellular domain of the protein. Short alleles, known as S (4 repeats), have less signaling efficiency compared to long alleles, known as L (7 repeats). A study of African-American smokers and non-smokers found a significant association between elevated risk of smoking and those individuals encoding the long allele (L). However, the same study analyzed a Caucasian population and did not find such an association24. The Caucasian population analysis revealed the association of the presence of the long allele (9 repeats) in the gene SLC6A3 (dopamine transporter) with an increase in the number of cigarettes smoked and the anxiety level25. In contrast, in Polish populations, those individuals who did not harbor the long allele in SLC6A3 had a higher risk of smoking before the age of 20, smoked a greater number of cigarettes daily, and had less abstinence compared to those carrying the short allele26.
The serotonergic system is involved in nicotine consumption. The serotonin transporter maintains serotonin concentrations in the nerve synapse, and the gene encoding for the pertinent protein (solute carrier family 6 member 4, SLC6A4) is a study candidate for addiction, given that it is related to addiction and depression. The promoter region of the gene is related to transcriptional efficiency and two common alleles - a 44-bp insertion (allele L) or deletion (allele S). Allele S has been associated with a decrease in transcriptional activity compared to allele L. In male Chinese smokers, genotypes L/L and S/L were found with a higher frequency compared to non-smokers. Moreover, such genotypes were associated with cigarette smoking and nicotine dependence27.
POLYMORPHISMS IN GENES RELATED TO NICOTINE ADDICTION IDENTIFIED BY GWAS
Using the GWAS strategy, genes involved in the addiction process not previously considered as functional candidates have been identified. Such is the case for neurexin-1 (NRXN1) and neurexin-3. The neurexin gene family encodes a group of cell-surface proteins that are mainly expressed in neurons; they are necessary for neurotransmitter release and are a key factor in synapse genesis. Three SNPs in the intronic region associated with nicotine addiction were identified in African-American and Caucasian populations. Due to their location, these SNPs will likely affect the alternative splicing of the mRNA, which generates isoforms that affect the formation of neural circuits28. Subsequently, rs1882296 in NRXN1 was associated with smoking risk in a Mexican population when comparing smokers to non-smokers. Using an in silico analysis, the presence of the risk allele (C) was observed to generate a miRNA capable of silencing genes that encode different receptors of the GABAergic and glutamatergic pathways compared to the presence of the common allele (T)29.
Vacuolar protein sorting 13 homolog A (VPS13A), homolog of VPS13A in Saccharomyces cerevisiae and brain-derived neurotrophic factor (BDNF) are examples of genes identified using the GWAS strategy. In VPS13A, SNP variants associated with a higher nicotine addiction risk have been found. In addition, variants of this gene have been observed to cause progressive neurodegeneration30. In a meta-analysis, genetic markers located in a region close to the BDNF gene were identified to be associated with tobacco consumption. This gene encodes proteins of the neurotrophin family which regulate the plasticity and survival of dopaminergic and cholinergic neurons. It is likely that variations in BDNF can alter the rewarding effects of nicotine through the modulation of dopamine reward circuits. On the other hand, an association between rs3025343 of the gene dopamine beta-hydroxylase and the cessation of tobacco use was observed; the protein that encodes this gene is involved in dopamine metabolism31.
One of the genomic regions with a higher replication in nicotine addiction is the group of genes formed by CHRNA5, CHRNA3, and CHRNB4. In 2009, Stevens et al. compared casual smokers (< 5 CPD) with heavy smokers (≥ 30 CPD) and identified 13 SNPs associated with the group of heavy smokers32. In the same year, a study on nicotine dependence in Caucasian and African-American populations included individuals with a low score (0-1) in the Fagerström test as the control group and individuals with a high score (≥ 4) as the nicotine-dependent group33. They found five SNPs with significant associations in CHRNA5, 11 in CHRNA3, and one in CHRNB4. In 2010, two SNPs associated with the number of cigarettes consumed in Korean populations were reported in the same genetic region34.
Variant rs16969968 in the gene CHRNA5 is the most replicated in association with nicotine dependence. This polymorphism causes a change in the amino acid sequence of aspartic acid (allele G) by asparagine (allele A) in position 398 of the protein (D398N), causing a change in the charge of the second intracellular domain of subunit α535.
Stimulation with acetylcholine, nicotine, or varenicline under the presence of asparagine in subunit α5 in cellular assays causes the nicotine-cholinergic receptors (nAChR) to respond more slowly compared to the subunit containing aspartic acid in position 398 (398N)36. The presence of allele A correlates with high expression levels in sputum and an increase in the risk of airway obstruction, even in non-smokers37. In vitro assays in lung epithelial cells show silencing of the expression of CHRNA5 with a decrease in the expression of adhesion molecules and an increase in cell migration capacity, contributing to airway damage38.
The decrease in the function of subunit α5 is associated with a significant risk of nicotine dependence; those subjects with allele A (398N) require a higher amount of nicotine to activate the dopaminergic pathway39. An example is a report in a European population, where individuals carrying the homozygous AA genotype are smokers consuming ≥ 20 CPD40. This change affects brain signaling in subjects who consume tobacco, causing significant changes in the ventral striatum, amygdala, and hippocampus regions41. Other reports indicate that smokers carrying variant A of rs16969968 are more likely to have strong memories associated with cigarette smoking, suggesting that allele A has an important role in the memory process42. In 2010, the tobacco and genetics consortium performed a meta-analysis of 16 original studies on nicotine dependence with some phenotypes associated with tobacco consumption; among their results, CHRNA5 polymorphisms were highlighted to be associated with cigarettes consumed per day, in particular, rs1696996831. Another important finding was allele A of rs1051730, which corresponds to an increase of 1 in the number of cigarettes smoked.
PHYSIOPATHOLOGY OF COPD
COPD is a broad term used to describe a group of lung disorders that share damage of the lung parenchyma (emphysema) and bronchial damage, which lead to the progressive obstruction of airflow. It is a multifactorial condition associated with exposure to inhaled toxic substances43. COPD has a progressive nature, with an increased presence of signs and symptoms over time, although it is known that the individual impact is quite variable. Likewise, only a fraction of smokers develops COPD, which highlights its multigenic nature44.
COPD is characterized by an abnormal chronic inflammatory response in the lung that causes irreversible structural changes. All smokers develop some level of lung inflammation, but in those developing COPD, the inflammation is more intense and harmful. Under normal conditions, humans can recover from the damage, and the healing process starts with an inflammatory response, important for the repair, and tissue remodeling processes. Inflammatory cells, mostly neutrophils, macrophages, and T CD8 lymphocytes, attract different mediators which remove the inhaled irritants. At the end of the process, the inflammation recedes. In contrast, in COPD, the airways remain continuously exposed to a great variety of toxic substances from the tobacco smoke, resulting in a continuous and amplified inflammatory reaction45. Cellular infiltrates are observed in the walls of large and small airways from the trachea to the alveolar ducts, including alveolar walls and blood vessels46.
Tobacco smoke exposes cells and tissues to high concentrations of oxidants and free radicals47, such as oxygen and nitrogen reactive species, which result in oxidative stress due to an imbalance with the natural antioxidant processes47,48. This oxidative stress directly contributes to the inflammatory process in the lung46, causing damage and cell death49. There is also an imbalance with proteases50, where cells such as neutrophils and macrophages release important mediators called proteases that are significant in mediating protection mechanisms. The natural balance to avoid the exaggerated action of these proteases is altered with the oxidative potential of cigarette smoke. Thus, endogenous antiproteases such as α1 antitrypsin (AAT), antichymotrypsin, and elafin oxidize and are inactivated, causing a protease-antiprotease imbalance. The excess of elastase not only causes emphysema but also promotes a higher production of mucins. Furthermore, tobacco smoke directly induces the death of lung cells (apoptosis)51. COPD damage starts in the respiratory epithelium, the portion most directly exposed to tobacco smoke and its cocktail of toxic chemicals52. Bronchial damage leads to chronic bronchitis, characterized by the presence of cough and chronic production of sputum. At the cellular level, there is a steep decrease in the ciliary apparatus in bronchial epithelial cells that predispose the individual to bacterial colonization, infections, and acute damage episodes called exacerbations53. There is also hyperplasia of mucus cells at the epithelial and submucosal gland levels. Other changes contributing to the chronic airway obstructions and air trapping include the development of peribronchial fibrosis and bronchospasm. Moreover, there is a gradual decrease in lung elasticity due to the progressive destruction of elastic proteins due to excessive protease. This destruction of the alveolar walls (emphysema) causes a marked reduction in the surface area necessary for gas exchange to occur.
COPD AND GENETIC SUSCEPTIBILITY FACTORS
Among the environmental risk factors, the most clearly associated is cigarette smoke exposure. Although the dose-response relationship between cigarette smoke and lung function is well established, there is an important variability in the level of airflow obstruction that occurs as a response to cigarette smoke54,55. The low percentage of the variation in lung function explained by cigarette smoke (approximately 15%), and the existence of early-onset cases with severely reduced lung function suggests variability in the individual genetic susceptibility to the effects of cigarette smoke55,56. In addition, it must be considered that only between 10% and 20% of patients with COPD are or were smokers57, indicating that there are other factors, including genetic, which confer susceptibility to develop the disease.
The deficiency of AAT (AATd) is considered a genetic risk factor, but it explains only < 1% of the cases57. On the other hand, a large variety of genes have been proposed as possibly involved in the development of COPD. These include α1 antichymotrypsin (SERPINA3)58-60, certain extracellular matrix metalloproteinases (MMPs) such as MMP161 and MMP962 and their inhibitor (tissue inhibitor of metalloproteinases 2)63, heme-oxygenase (HMOX1)64, microsomal epoxide hydrolase (EPHX1)65, glutathione S-transferase p1 (GSTP1)66, and glutathione S-transferase m167, lung surfactant proteins b and d (SFTPB)68 and SFTPD69, Vitamin D-binding protein (GC)70, interleukin (IL) 1371, tumor necrosis factor α (TNF-α)72-74, C-C motif chemokine ligand 174, decorin, transforming growth factor β1 (TGFβ1)74, and the adrenergic receptor β2 (ADRB2)75 as some examples. The previously established associations of individual genes include biological pathways with diverse functions, including immune response, inflammation, xenobiotic metabolism, antioxidants, and protease/antiprotease systems76.
Studies based on families have demonstrated an increase in the risk of lung function damage in the first-degree relatives of smoking or ex-smoking patients55. Lung function studies in twins provide additional evidence on the role of genetic factors in determining lung function in the general population77. Moreover, since the 1970s, several authors have reported an increase in the prevalence of airway obstruction among family members (first-degree relatives) of patients with COPD compared with the family members of control subjects78. In addition, the existence of variability in lung function measurements has been described, with a high heritability rate79 as well as severe and early-onset forms55.
Recently, six new loci (in or close to EFEMP1, BMP6, MIR129-2-HSD17B12, PRDM11, WWOX, and KCNJ2) have been associated with forced vital capacity (FVC) in a population of Caucasian descent, and two loci previously associated with spirometry measurements (GSTCD and PTCH1) related to FVC were identified using a GWAS meta-analysis, achieving the detection in human lung tissue of transcripts of all six genes recently involved80. Such loci could be implicated in lung development mechanisms and in the pathogenesis of restrictive lung disease. Two new loci (IL16/STARD5/TMC3 on chromosome 15 and ME3 on chromosome 11) were previously identified and associated with the forced expiratory volume in the first second (FEV1) change rate that contains candidate genes with a biologically feasible involvement in lung function81.
POLYMORPHISMS IN GENES ASSOCIATED WITH COPD SECONDARY TO TOBACCO SMOKING
Polymorphisms found within the SERPINA1 gene, encoding AAT, are among the genetic factors that are clearly associated with the development of COPD. It is a highly polymorphic gene, with > 100 variants of the protein described, of which 30 could have a pathological consequence82. Protein variants are classified according to their electrophoresis migration velocity in a magnetic field with different pH gradients. Pioneering researchers in this area designated these proteins as M (medium) for those with medium velocity, F (fast) for those with fast migration, and S (slow) for those with slow migration83. When new variants were discovered, the anodic proteins were assigned the first letters of the alphabet, while the cathodic ones were assigned the last letters82. The normal genotype, present in > 90% of healthy individuals (94-96% in Caucasian populations), is called PiMM and is characterized by serum levels of approximately 150-350 mg/100 mL. Variants S (Glu264Val) and Z (Glu342Lys) constitute 95% of the mutations in patients with severe AAT deficiency; both are nonsense mutations in the SERPINA1 gene (rs17580 and rs28929474, respectively). Individuals with genotypes SS, SZ, and ZZ express serum concentrations of the protein of 85%, 25%, and 15%, respectively, compared to the normal levels (genotype MM)84.
Most studies conclude that genotype SZ is less important than ZZ because patients with genotype SZ develop emphysema at an older age compared with those carrying ZZ82. Alleles S and Z encode abnormal proteins that polymerize in the liver, and therefore, 80-90% of PiZ and 40-50% of PiS molecules are retained inside the hepatocyte-forming polymers, usually degraded by the proteasome. In clinical practice, the risk of having diseases related to AAT deficiency is limited to the phenotypes of ZZ (96%)82. The progressive decline of lung function in homozygous ZZ individuals is well known; regarding the medium deficiency genotypes (MZ and MS), it was determined that, compared to genotype MM, MZ is associated to a reduction in lung function in individuals with COPD. Genotypes SZ and ZZ are associated with airway obstructions and lung function reduction, particularly in smokers85,86.
The prevalence of these polymorphisms is < 5.0% (0.2% for PiZ and 3.42% for PiS) in Mexican Mestizo population. However, the presence of the heterozygous variant PiS (AT) is associated with lower FEV1/FVC ratio compared with those individuals who do not have this polymorphism87.
Other genes associated with COPD that contributes in small proportions to this pathology include those encoding the IL 6 receptor (IL6R) and glutathione S-transferase omega 2, associated with decreases in lung function and/or COPD79.
Then, based on the known fact that lung function measurements are hereditary traits predicting morbidity and mortality related to COPD development88, the cohort of the SpiroMeta consortium (European descent individuals) evaluated the association with lung function measurements (FEV1 and FEV1/FVC). The authors confirmed the formerly reported association between locus 4q3189, previously associated with lung function, and COPD79.
Interestingly, protective alleles have also been described in Hispanic populations in the USA90, a finding that has been replicated in Mexican populations in the genes IL6R and ADAM1991.
GWAS IN THE STUDY OF COPD
Multiple studies have evaluated the individual involvement of genetic markers, mainly SNPs, in COPD susceptibility, while few include whole genome searches for new variants to find alternative or novel hypotheses for the underlying mechanisms of the pathology.
In the Framingham Heart Study, a GWAS was performed to identify genetic markers related to lung function measurements, identifying SNPs in two genes not previously described using traditional methods associated with lung function measurements. In this study, participants were grouped by phenotype according to the spirometry results. The authors suggested that, for SNPs with significant results, especially those located in genes with unknown biological function, a replica in another group of individuals is required79. Pillai et al. reported, in 2009, a GWAS in COPD where they used the multiple stage replication technique. Initially, in the study, 538,030 SNPs were analyzed in a group of cases and controls, obtaining a list of associations, from which the 100 SNPs (top 100) most strongly associated (p = 1.6E-04)) were selected. These SNPs were characterized in an additional cohort of families, which they labeled replication Stage I. Based on the results from replicate I, the seven SNPs with the best association evidence in the joint analysis of both stages (p < 1.0E-07) were selected and analyzed in an additional case and control population, labeled replication Stage II. Finally, they performed a study for the validation of results in a cohort of individuals with COPD, resulting in six validated SNPs with a significant association to the disease. It is important to note that they found genes that had not been previously associated with COPD but that play a key role in other pathologies such as lung cancer. Such is the case of the CHRNA3/5 cluster. Another gene highly associated with COPD is HHIP, about which little is known, but the authors suggest that, based on their results, it should be extensively studied92. Likewise, 70,978 SNPs were analyzed in a GWAS related to lung function measurements in the Framingham study cohort. Their results propose the HHIP gene as one of the possible regulators in COPD and suggest the importance of the gene encoding glycophorin A (GYPA) in the disease due to its position in relation to HHIP and earlier findings of this protein in patients with COPD. Regarding the gene CHRNA3/5, they did not obtain significant results93.
Recently, five loci associated with different lung function measurements were identified using GWAS associated with lung function measurements (FEV1 and FEV1/FVC)89. In addition, the authors performed a meta-analysis with the strongest association signals from direct genotyping, a summary association of in silico data of the CHARGE consortium, and ultimately included data from the Health 2000 study, confirming the previously reported association of locus 4q31 as well as the associations with FEV1 and/or the FEV1/FVC ratio and common variants in five loci. The analysis of mRNA showed expression of TNS1, GSTCD, AGER, HTR4, and THSD4 in human lung tissue. Finally, they suggest that these associations offer signals of the mechanisms that regulate lung function and indicate potential targets for therapeutic interventions in respiratory pathologies89. In general, 17 regions were observed to be associated independently with FEV1, and 23 were associated to the value of the FEV1/FVC ratio, including three regions (4q24 in GSTCD, 4q31 near HHIP, and 15q23 in THSD4). The SNP rs12504628 was found to be associated with both FEV1/FVC and FEV1 traits; it is located in an intergenic region close to HHIP, covering ~300 kb of the 4q31 region that was previously associated with lung function and COPD93. The hedgehog gene family, of which HHIP is a member, encodes molecules involved in the regulation of lung morphogenesis, suggesting other overlying mechanisms for such associations94.
In parallel, a study analyzing lung function measurements was published using a meta-analysis-like design and GWAS. This study included 20,890 participants of European descent (the same used in a study by Repapi et al.89, defining eight loci associated with the FEV1/FVC ratio (HHIP, GPR126, ADAM19, AGER-PPT2, FAM13A, PTCH1, PID1, and HTR4) and one locus associated with FEV1 value (INSTS12-GSTCD-NPNT)94,95. Several SNPs near the HHIP coding region were associated with the FEV1/FVC ratio in both the CHARGE and SpiroMeta consortia, confirming previous findings performed in the Framingham Heart Study79.
THE CANDIDATE GENE STRATEGY IN COPD
On the other hand, association studies using the candidate gene strategy in cases and controls are frequently criticized due to the lack of population replicates96,97, even finding contradictory results in some cases or the absence of association with previously identified genetic markers98. In this regard, Hersh et al. performed a replication study in 2005 based on families and in parallel on cases and controls using the candidate gene strategy and individual genetic associations related to COPD development, where 29 polymorphisms were evaluated (24 SNPs, 1 insertion/deletion [indel], 3 STR, and 1 nonsense deletion) in 12 candidate genes previously reported on the literature. They found significant associations related to distinct qualitative and/or quantitative phenotypes with different SNPs, including TNFα (−308G > A), SFTPB (thr131ile), HMOX1 (gt31), and EPHX1 (his139arg)76.
GENES RELATED TO INFLAMMATION AND THEIR ROLE IN COPD
Different mediators and cells have been involved in the pathology of COPD, including TNFα, IL-8, and TGF-β. Cigarette smoke activates macrophages that release TNFα, leukotriene B4, IL-8, and other neutrophil chemotactic factors, as well as antiproteases. This reaction increases the local inflammation, with a predominance of neutrophils and protease release99. IL-8 is a chemokine that mediates the activation and migration of neutrophils from peripheral blood into the tissue. It plays a significant role at the onset of the amplification of the inflammatory response. A transversion (A→T) in position −351 and another in −251 of the IL-8 promoter have been associated with COPD in different populations100,101 but are inversely associated with bronchial asthma102,103.
An SNP was previously described in the promoter region within the TNF gene that was identified because it directly affects the transcriptional regulation of the gene104. Several studies show certain relevance for the SNP −308G/A of TNF in Asian populations but not in Caucasian ones. For example, in Japanese and Taiwanese populations, an increase in the prevalence of COPD has been observed compared to their respective control groups72. However, these results have not been confirmed in other populations105,106, and given the location of the TNF gene within the HLA region, it would be important to establish the existence of extended haplotypes that include the classic HLA genes. Recently, our group identified certain genetic variants associated with both the susceptibility and severity of COPD related to smoking107 and smoke exposure due to the burning of biomass108.
The expression of HLA-DR and CD80 in the cell surface of alveolar macrophages (AM) in patients with COPD is decreased compared to smokers with normal lung function and to non-smokers. In addition, these patients show an increased percentage of AM with low levels of expression of CD44 on the surface109. Genes encoding HLA antigens, especially class II, are particularly interesting to us.
ASSOCIATION BETWEEN HLA ALLELES AND COPD
Despite the contribution of allelic variation of the HLA system to several diseases, mainly autoimmune and inflammatory, the role of HLA alleles in COPD has not been extensively studied; only a limited number of studies have described this association110. Among the few reports on the association of this disorder with the HLA system, the 1990 study by Sugiyama et al. typified antigens HLA-A, HLA-B, and HLA-C using serological methods in patients with diffuse panbronchiolitis. This is a form of COPD with unknown etiology, which is clinically characterized by chronic cough, sputum, and dyspnea, physiologically by the limitations of airflow, and histologically by inflammatory lesions around the bronchioles, with an increase in the frequency of HLA-B54 antigen111. Although the number of patients in this study was relatively small, it is important to mention that HLA-B54 is a serotype mainly found in East Asians. Subsequently, Keicho et al. discovered, in 1998, a positive association between PBD and alleles HLA-A11, HLA-B54, and HLA-Cw1, while HLA-A33 and HLA-B44 increased their frequencies in the control groups, suggesting a protective association for these alleles in Japanese populations112. In addition, the findings for A11 were replicated in Korean populations, but those for B54 and Cw1 were not; in contrast, HLA-B55 is increased, and HLA-B62 and HLA-Cw4 are weakly associated113. Regarding Caucasian populations, the oldest record in the literature is the one from Kauffmann et al., who in 1983 demonstrated that HLA-B7 is a genetic risk factor for developing COPD; the finding is worthy of mentioning given that HLA-B7 is predominant in Caucasian populations114. Kasuga et al., in 2005, performed a replication study where the tentative association between HLA-B7 and COPD described by Kauffmann was analyzed using a casecontrol design in smokers. They studied the contribution of HLA-B7 to the decay of lung function, which was measured by FEV1, but they did not find significant differences in HLA-B7 frequencies between COPD patients and patients without airway obstruction nor an association between HLA-B7 and lung function decline. These findings allow us to conclude that HLA-B7 does not contribute to COPD development and is not involved in lung function variations110.
GENES OF EXTRACELLULAR MMPs AND COPD
MMPs are a family of at least 20 proteolytic enzymes that have an essential role in tissue remodeling. Most MMP genes contain hundreds of polymorphisms115, some of which are associated with COPD. At least 20 polymorphisms in MMP1 gene have been associated with COPD116, mainly in regulatory regions (promoter, 3UTR, 5UTR, and introns)117. Some genetic studies associating MMPs with COPD include genes such as MMP1, MMP2, MMP3, MMP9, and MMP12.
The insertion/deletion polymorphism 1G-1607/2G in the promoter region of MMP1, which increases transcription due to the introduction of a new binding site for the ETS-1 transcription factor118, has been evaluated in relation to lung function decline, with controversial results119,120. In Russian population, the homozygous genotype 2G was associated with risk in COPD patients121, a finding that was not replicated elsewhere121,123. In addition, rs1799750 in MMP1 was identified as associated with the mainly apical densitometric distribution of emphysema124, while the carriers of allele T of rs470358 are associated with the transference of gases in patients with COPD secondary to AATd125. Finally, polymorphisms in MMP1 have also been associated with the predisposition to occupational chronic bronchitis126.
In MMP2, the homozygous genotype T of the polymorphism rs243865 (C-1306T), located in the intron, shows a significant association with the FEV1 decrease in Caucasian populations127, while in Tunisian populations, it was correlated with disease severity128. In Mexican populations, rs243864 and rs11646643 were associated with the risk of COPD, and in addition, MMP-2 serum levels were lower129.
The identification of MMP-3 in COPD is recent. At the genetic level, it has been described that, in patients with AATd, those carrying allele rs678815/G in MMP3 shows a lower transference of gases125. Genotype 6A6A of polymorphism −1171 5A > 6A (rs35068180) was associated with a significantly high risk of COPD in Russian population123 and with COPD lung cancer in Polish population130. Polymorphisms rs3025058 and rs678815 have been proposed as susceptibility markers for the development of lung cancer in individuals with COPD131.
Another well-explored gene in this metalloproteinase family is MMP9. Polymorphism rs13925 (C-1562T), located in the genes promoter, is associated with COPD development in Japanese62 and Chinese populations132,133 and was recently confirmed in a meta-analysis including Caucasian and Asian populations134. In Russian populations, allele T and the homozygous genotype TT are associated with increased severity and younger age of disease onset122. In Northern European populations, rs3918242 was associated with centrilobular emphysema134. In Tunisian populations, a significant correlation was identified between polymorphism 279 R/Q (836G > A) and disease severity. Likewise, Q homozygotes (genotype AA) were associated with a drop in FEV1 and FVC in patients with COPD compared to individuals with genotypes AG and GG. In contrast, the activity of MMP-9 improved in individuals carrying allele R (G) compared to those homozygous for variant Q (A)135. Our group used SNP tagging and determined that the rs3918253 of MMP9 associated with COPD risk increased in serum levels in a group of patients with stable COPD129.
Polymorphism rs2276109 (−82A/G) of gene MMP12 was associated with an increase in the promoter activity and has an effect in the cis element of transcription factor AP-1136. It is also associated with a positive effect on lung function in children with asthma and adult smokers. This allele is further associated with a reduced risk of COPD in adult smokers137. Although only one tendency was identified in Polish populations130, in Bulgaria, patients carrying genotypes with at least one copy of allele G could have a lower risk of COPD138.
Recently, it has been proposed that COPD has its origin in early childhood, by demonstrating associations between several important genes related to COPD and early transient symptoms (wheezing and decrease in lung function). The associated genetic variants include rs2276109 in MMP12137. In Indian populations, this polymorphism showed an association in additive and dominant models, while the infrequent allele (G) showed a significant positive association with lung function in different genetic models. In contrast, haplotypes carrying allele A showed a negative correlation with lung function139. Genetic associations have also been described for SNPs in or near MMP12 with different lung emphysema patterns quantified by automated tomography140. This polymorphism has been demonstrated to be associated with the severity of lung disease141. Recently, with the advent of exome analysis, the associations of SNPs rs17368582 and rs2276109 could be replicated142. Polymorphism Asp357Ser (rs652438) was associated with the decline rate in lung function in Caucasian smokers61 and with COPD in Chinese populations143. The common variant (Ser) in codon 357 is associated with clinical manifestations that are consistent with a more aggressive matrix degradation in other tissues; it is associated with an increased severity of the disease in patients with COPD137.
GENETIC CONSIDERATIONS IN COPD EXACERBATIONS
Hospitalizations are an important part of patient care and are directly related to the deterioration of the health status144, mortality increase145, and a substantial rise in health-care costs146. Hospital readmissions are common and occur in an alarming 60% of patients in the 1st year after the last exacerbation147,148.
At the molecular level, a reduction in the expression of HLA-DR on the surface of peripheral-blood mononuclear cells has been described, as well as a higher frequency of allele QBP 5.12 in the promoter of the gene HLA-DQB1 in patients under exacerbation149.
Infectious exacerbations are common in patients with COPD, where cells and molecules of the immune response are involved at different levels. The protein mannose-binding lectin (MBL) is a key component in the defense of the host against an infection150. It is a pattern recognition receptor that binds to carbohydrates on the microorganisms surface, which, in turn, activates the complement system151,152. The protein MBL is encoded by the gene MBL2; patients carrying allele D in codon 54 of exon 1 of MBL2 (rs1800450 G/A), which at the protein level consists of a substitution of glycine (G) by aspartate (D), have more risk of hospitalization due to infectious exacerbation153. In recurrent exacerbations, the mortality and frequency of infectious exacerbations were significantly higher in patients with deficient MBL genotypes than in those with non-deficient MBL genotypes154.
Lung surfactant is a multimolecule complex formed by phospholipids and cholesterol (90% in total) and surfactant proteins (SPs) (10%); multiple proteins of its composition have been associated with respiratory pathology. Among them, those known as lung SPs can be found. They are composed of the high molecular weight hydrophilic proteins SP-A and SP-D; the extraordinarily lipophilic, low-molecular weight SP-B; and the SP-C, which are vital for the biophysical properties of surfactant phospholipids155. SP-D has immunomodulatory functions156; by keeping a potentially aggressive inflammatory reaction under control, SP-D decreases the production of proteases and oxidants157. The association between rs3024791 in SFTPB (SP-B) and COPD exacerbation was confirmed using logistic regression models. Negative binomial regression models demonstrated the association of multiple SNPs (rs2118177, rs2304566, rs1130866, and rs3024791) with exacerbation rates158.
Sialic acid-binding immunoglobulin-like lectins (Siglecs) are a group of cell-surface transmembrane receptors expressed on immune cells that regulate the immune equilibrium in inflammatory diseases159. Homozygotes for the null allele in SIGLEC14 are associated with a reduced risk of exacerbation in a population of Japanese patients160, while allele G of rs2075803 and haplotype GA (rs2075803 G/rs2258983 A) in SIGLEC9 were associated with a higher frequency of exacerbations and with the emphysema level161.
Using site-directed mutagenesis and recombinant expression studies, it was found that several polymorphisms of the gene ADRB2 alter the function of ADRB2. Of these, polymorphisms in positions 16 (Arg16Gly) and 27 (Gln27Glu) are prevalent, are functionally relevant, and have been widely studied from the clinical and pharmacogenetic perspective.
The homozygous genotype Arg16Arg predisposes patients to a clinically more severe episode162, greater dyspnea, more symptoms, and frequent exacerbations163. Patients with genotype Arg16Arg in ADRB2 showed better results during exacerbation in response to salmeterol than genotypes Gly16Gly and Arg16Gly, which suggests a potential differential effect of genotype Arg16Gly in the response to the treatment with long-acting β-agonists164.
Haplotype Gly16/Glu27 can affect the severity of the obstructive ventilatory alteration but not the immediate response to salbutamol during the acute exacerbation165. On the other hand, two independent pharmacogenetic studies in patients with moderate-to-severe COPD showed that the therapeutic response and the tolerance to long-term treatment with formoterol (alone or in combination with budesonide) were not modified by the genotype of Arg16Gly166. Another receptor involved in the pharmacological response is the muscarinic cholinergic receptor 2 (CHRM2). Recently, a significant association of polymorphism rs1824024 in CHRM2 with disease severity was identified, with lower values in lung function tests, frequent exacerbations, and a poor response to anticholinergic drugs167.
Other agents associated with COPD exacerbations include biological pathways such as the metabolism of xenobiotics, glucocorticoids, and inflammation. GSTP1 (rs1695), superoxide dismutase 2 (SOD2) (rs4880), and NFE2L2 (rs1806649) were associated with a tendency toward a higher risk of hospital admissions during the periods with high PM10 levels154.
Individuals with allele C for rs4588 in the GC gene showed a higher exacerbation frequency, higher susceptibility to COPD, emphysema, and a tendency toward a rapid decrease of airflow obstruction168. The minor allele of SNP rs2227744G > A in the protease-activated receptor 1 (F2R) was associated with protection against frequent exacerbations169, while genotype DD of the gene ACE was associated with a lower risk in males170.
GENETIC POLYMORPHISMS RELATED TO NICOTINE ADDICTION AND COPD
Polymorphisms associated with nicotine addiction and COPD have been identified. Among genes with such pleiotropic effects are those encoding nAChR, specifically subunits α5 and α3 (CHRNA5-CHRNA3)171,172. These genes not only encode subunits that can be part of the neuronal nAChR but also form part of the muscle receptors.
The nicotine that enters the body of the smoker activates these receptors in the brain, unleashing the signaling for the release of neurotransmitters, with dopamine being the most important in the addiction process. This increase in dopamine in the brain acts as a positive reinforcer, causing the constant need to smoke cigarettes173,174. It has been observed in vitro that nicotine reversibly induces the expression of CHRNA5, which could be contributing to the nicotine addiction mechanism175. The non-neuronal nAChR that contain subunits formed by proteins encoded by CHRNA3 and CHRNA5 are expressed in airway cells (bronchial epithelial cells, neutrophil macrophages, monocytes, and lymphocytes) and are also activated when nicotine enters the lungs, causing the release of proteases, pro-inflammatory cytokines, and oxidants that are responsible for lung remodeling, which leads to lung damage after chronic exposure176 (Fig. 2).
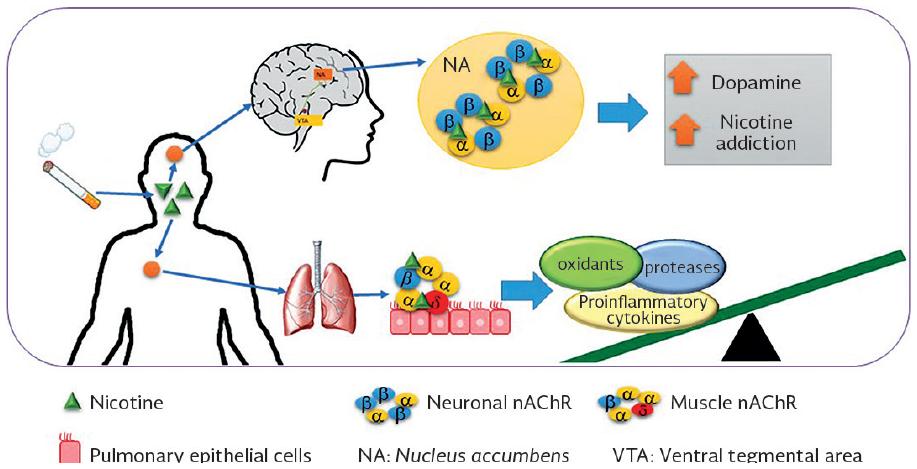
Figure 2 Proposal of the dual participation of nicotinic cholinergic receptors in nicotine addiction and chronic obstructive pulmonary disease.
SNP rs16969968 in CHRNA5 is among the SNPs localized in genes encoding nAChR with the highest evidence of association with nicotine addiction, CPD, and COPD. A meta-analysis including 34 studies (case-control studies, families, and cohorts) reported that allele A of this SNP was associated (p = 5.96E-31) with a higher risk of cigarette smoking in European populations when comparing heavy (CPD > 20) versus casual smokers (CPD ≤ 10)177. This polymorphism was also associated with airway obstruction independent of cigarette smoking in seven European population cohorts37, and when Asian populations are analyzed, the association with COPD remains when the risk allele (A) is present.
Other interesting polymorphisms in the cluster of genes in CHRNA3 are rs6495309 (T/C) and rs1051730 (C/T). The latter was evaluated in smokers and non-smokers in Denmark; in smokers, the presence of genotype TT was associated with the risk of suffering from COPD in more severe stages (GOLD III-IV)178. In a Dutch cohort of smoking individuals, it was reported that carriers of allele T (rs1051730) showed an increased risk of presenting bronchial obstruction and emphysema179. It has been reported that carrying a copy of allele T increases smoking by 1.2 CPD, suggesting that this region of chromosome 15 has an important function in nicotine levels in smokers180. On the other hand, for rs6495309 in populations from Southeast China, the presence of allele C was reported to be a risk factor for developing COPD and leads to a higher annual decrease in FEV1. Later, cellular assays suggested that the presence of this allele increases the transcriptional activity of CHRNA3181.
In a Russian population that included patients with COPD and clinically healthy individuals (26% non-smokers), it was found that the haplotype formed by alleles AAG in CHRNA3/A5 (rs16969968-rs1051730-r6495309) was present at a higher frequency in patients with COPD than in the control group182. In Mestizo Mexican populations, we have identified, using multi-stage association studies, polymorphisms in CHRNA5 (rs16969968 and rs17408276) that are associated with cigarette smoking29. A later analysis, including smokers with and without COPD, showed that patients with COPD and of Caucasian descent had a haplotype formed by 12 SNPs in CHRNA5/A3 (AGGAAGAGGGCA) that includes rs16969968 and rs17408276. This group of genetic variants was strongly associated with the risk of suffering from COPD; the same behavior is not reported in those individuals of mostly Amerindian ancestral descent183.
AGING AND COPD
With increasing age, even in healthy individuals, lung function declines and the respiratory system also experiences structural changes that include pulmonary remodeling, limited tissue regeneration, and an enhanced susceptibility to pulmonary diseases184. Age-related alterations in the respiratory tract originate from cumulative damage or oxidative stress occurring throughout life. The impaired DNA repair capacity leads to accumulation of oxidative damaged DNA and cell senescence185. There is increasing clinical and cellular evidence for the concept that accelerated aging can serve as an underlying mechanism of COPD186. An accelerated cell senescence in alveolar Type II epithelial cells has been observed in COPD patients187. Moreover, telomere length dysfunction, decreased expression of anti-aging molecules such as sirtuins, increased expression of senescence-associated molecules including p16 and p21, and β-galactosidase activity have been observed in lungs of patients with COPD187-190.
Telomere shortening, one of the proposed nine hallmarks of aging191, has been implicated in a variety of lung diseases such as idiopathic pulmonary fibrosis, COPD, emphysema, and lung cancer192-194. Telomeres are the protective structures at the end of chromosomes that consist of stretches of repetitive hexanucleotides (5-TTAGGG-3). They protect the end of chromosomes from erosion or fusion by DNA repair processes. Telomeres become progressively shorter as cells divide, due to an event that is known as the end-replication process195. The enzyme telomerase is capable of maintaining telomere regeneration, but its activity is limited mainly to embryonic and adult stem cells. Telomeres in regular somatic cells are therefore reduced with each replication until critical telomere length is reached leading to cell cycle arrest185.
Many studies have tried to shed some light on the interaction between telomere length and COPD. Shortened telomeres have been described in current and former smokers in comparison with non-smokers196. Several studies have documented shorter telomeres in circulating leukocytes in patients with COPD than age-matched controls197-199. Interestingly, shortened telomeres measured in peripheral leukocytes of patients with COPD have been related to all-cause and cancer mortality200 supporting the concept that telomere length could serve as a biomarker of disease progression. Recently, the first longitudinal study on telomere dynamics and COPD has shown an accelerated telomere shortening in patients compared to age-matched smoker controls over a 3-year period of follow-up201.
However, what do we know about telomere attrition and its relationship with lung function in COPD? The results from different studies are controversial. A study performed over a large cohort of patients with COPD has reported a weak correlation between telomere length and the lung function expressed by the FEV1202. Others have shown that leukocyte telomere length was the only marker between a panel of aging markers that were associated with lung function (FEV1)203. However, the absence of a relationship between telomere shortening and lung function has also been described197,199. A meta-analysis performed over seven studies has found just a modest association between telomere length and FEV1 decline204. In the same line, a recent study that analyzed other parameters (inspiratory capacity/ total lung capacity, KCO, and PaO2) besides FEV1 could not find any association between them or the change in lung function over time and telomere length201. Of note, if present, the association of telomere attrition with lung function would not be so strong. Comparisons between different studies are difficult because of many factors that may influence the results, for example, the cohort sample size, diverse stages of disease severity, the proper correction for confounders, or the technique used to measure telomere length.
To expand our knowledge over the role of telomere biology in COPD and to explore about causality, studies on large cohorts of patients with telomere length measurements at multiple time-points are needed. Unraveling the molecular mechanisms that link premature aging with COPD would allow the development of promising antiaging therapies with impact on disease progression and outcomes in COPD.
SOCIAL IMPACT OF COPD AND ITS RELATIONSHIP WITH GENETICS
Signs and symptoms such as dyspnea, persistent cough, wheezing, sputum, poor tolerance to exercise, weight loss, and fatigue are typical indicators of COPD205, in addition to anxiety and depression148,206,207. Depression as a response to hypoxia in patients with COPD can be influenced by genetic factors, with an autosomal dominant inheritance model208. On the other hand, the severity of depression is higher in patients with COPD than in smokers without COPD. Genotype Val/Val at position 9 of the signal peptide of the SOD2 enzyme is associated with more severe depression, trait anxiety, and state anxiety compared to patients with genotypes Val/Ala and Ala/Ala209. In Japanese populations, rs3794808 in SLC6A4 was correlated with the Hospital Anxiety and Depression Scale score210.
CONCLUSION
There are numerous studies that provide evidence of the involvement of a genetic component that contributes to the risk of developing nicotine addiction and COPD. In addition to genetic variability as a component in the susceptibility in its simplistic way, this contribution could be associated to clinical parameters. These include severity, age of onset, increased tobacco intake (as CPD, heavy consumption, high addiction, etc.), the decline of lung function, frequency of exacerbations, alterations in protein levels driving to chronic inflammatory, and/or oxidant processes, often accompanied by histopathological changes mediated by cellular mechanisms easily altered by tobacco smoke and consequent epigenetic changes. This multifactorial nature of nicotine addiction and COPD requires coordinated research from multiple disciplines, moving forward to precision medicine.

