











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
Palladium(II) complex K2PdCl4 has attracted considerable attention due to the medicinal properties as well as applications in drugs with high antitumoral and antibacterial activity1.Meanwhile, K2PdCl4 can act as a catalyst to afford an efficient, mild and environmentally friendly method in the synthesis of
Usually, the above properties and applications may strongly depend upon the electronic structures and local behaviors of Cr3+ in these systems, and the investigations on the local structures and behaviors of Cr3+ in K2PdCl4 are of scientific and practical significance. It is known that EPR spectroscopy is sensitive to the immediate environment around the paramagnetic impurities and able to reveal important microscopic information (e.g., defect structures such as occupation, strength of crystal-fields and local lattice distortions etc.) in the hosts. Recently, electron paramagnetic resonance (EPR) study was carried out for Cr3+ doped K2PdCl4 at liquid nitrogen temperature (LNT), and the spin Hamiltonian parameters (SHPs) were measured for the orthorhombic substitutional Cr3+ centre on K+ site (site I)14. So far, the above EPR experimental results for Cr3+ doped K2PdCl4 have not been theoretically explained, except that the d-d transition optical spectra were quantitatively analyzed based on the crystal-field theory [14]. Since this orthorhombic Cr3+ centre on the host K+ site in K2PdCl4 is quite different from the conventional tetragonal Cu2+ centre on square planar Pd2+ site, further theoretical investigations on the SHPs of this system can be helpful to understand the impurity behaviors and the properties of K2PdCl4 with transition-metal dopants and are of specific importance. The aim of this article is to propose satisfactory and uniform interpretations of the SHPs for the Cr3+ centre in K2PdCl4 by involving the local structure of the impurity based on the perturbation calculations of these parameters. The arrangement of this paper is the follows. In Sec. 2, the relevant theoretical formulas for the calculations of the SHPs are provided. The results are discussed in Sec. 3. The conclusion of the article is drawn in the last section.
2. Theoretical calculations
In this section, the theoretical calculations of the SHPs are performed for the Cr3+ centre in K2PdCl4. In the treatments, the contributions to the SHPs from both the crystal-field (CF) and charge-transfer (CT) mechanisms are included in view of significant covalency of the system. EPR measurements14 indicate that Cr3+ in K2PdCl4 occupies substitutionally the host K+ site and exhibits the local orthorhombic (D2h) site symmetry. For a 3d3 (Cr3+) ion under orthorhombically distorted octahedra, its ground orbital singlet A2g may yield ZFSs D and 𝐸 via the combination of the spin-orbit coupling and the orthorhombic CF interactions15.
2.1 Cluster approach treatments involving both the CF and CT contributions
In view of the high valence state of central ion Cr3+ and the strong covalency of ligand Cl-, the studied CrCl6 cluster may demonstrate strong covalency and CT contributions to the SHPs15-17. Based on the cluster approach18,19 involving both the CF and CT contributions, the spin-orbit coupling coefficients and the orbital reduction factors are expressed as follows:
 (1)
(1)
Here
2.2 Perturbation formulas for an orthorhombically distorted 3d3 cluster
For this strongly covalent system, not only the contributions from the conventional CF mechanism (related to the anti-bonding orbitals) but also those from the CT mechanism (related to the bonding and non-bonding orbitals) affect significantly the 𝑔-shifts arising from the second-order perturbation term due to the combination of spin-orbit coupling and orbital angular momentum interactions18,19. As for ZFSs D and E, the CF contributions originate from the third- and fourth- order perturbation terms due to the combination of orthorhombic CF and spin-orbit coupling interactions or the off-diagonal parts of the inter-electronic repulsion interactions, while the CT ones arise mainly from the third order perturbation terms due to the combination of orthorhombic CF and spin-orbit coupling interactions. Utilizing the Macfarlane’s perturbation-loop method20,21 and involving the CT contributions to 𝑔 factors and the ZFSs 𝐷 and 𝐸, one can obtain the improved perturbation formulas of the SHPs for an orthorhombically distorted octahedral 3d3 cluster as follows:
 (2)
(2)
The quantities Ds, Dt, 𝐷 𝜉 and 𝐷 𝜂 are the orthorhombic CF parameters. The corresponding denominators E i (I = 1 - 4 and 8)20,22 are the energy differences between the ground 4A2g and the excited 4T2g [t 2 2(3T1)e], 2T2g (t3 2), 2T2g [t2 2 (3T1)e], 4T1g[t2 2 (3T1)e] and 2T1g [t2 2 (3T1)e] states, respectively. These denominators are expressed in terms of the cubic CF parameter Dq and Racah parameters B and C from the strong field approach23,24:
For an octahedral 3d3 cluster, the CT level can be determined as
3. Calculations for the orthorhombic Cr3+ centre in K2PdCl4
The host K+ site in K2PdCl4 is surrounded by a tetragonally compressed chlorine octahedron, with the cation-anion distances

Utilizing the local geometry and the superposition model27-31 the orthorhombic CF parameters can be determined as:
Here
In the light of the optical spectral measurements for Cr3+ in
K2PdCl414 and similar chlorides35, the cubic field parameter
The corresponding theoretical SHPs (Calc.c) are collected in Table 1. The results (Calc.a) based on the host structural data of K3+ site in K2PdCl4 and inclusion of both the CF and CT contributions and those (Calc.b) based on the above optimal local distortion parameters and inclusion of merely the CF contributions are also listed in Table 1.
Table 1 The ZFSs D and E (in 10¡4 cm¡1) and g factors for Cr3+ in K2PdCl4.
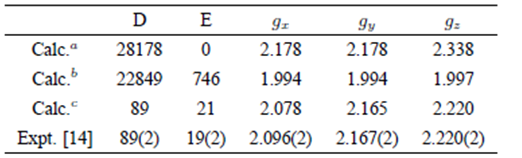
aCalculations based on the host structural data of K+ site in K2PdCl4 and inclusion of both the CF and CT contributions.
bCalculations based on the optimal local distortion parameters
cCalculations based on the optimal local distortion parameters
4. Discussion
Table 1 indicates that the theoretical SHPs (Calc. c) based on the local distortion parameters 𝜌 and 𝜏 show better agreement with the experimental data than those (Calc. a) based on the host structural data of K+ site and those (Calc.b) based on omission of the CT contributions. Thus, the measured EPR spectra for the Cr3+ centre in K2PdCl4 are satisfactorily explained in a uniform way. Meanwhile, the information about the local distortion around the impurity Cr3+ is theoretically determined by analyzing the experimental ZFSs. The following points are discussed here.
(1)As compared with the axial compression ratio (
(2)When the CT contributions are ignored, the calculated g factors and ZFSs (Calc. b) are smaller and much larger than the experimental data. For the studied [CrCl] group, the system demonstrates strong covalency due to the covalent ligand Cl- and central ion Cr3+ with high valence state and much larger ζp0(Cl-)
(3)The errors of the present theoretical calculations may be analyzed as follows. First, the final results may be brought forward some errors due to the approximations of the theoretical model and formulas based on the spin Hamiltonian theory, in which only the contributions from the nearest neighbor ligands are considered whereas the influences of the ions in the outer ligand spheres are omitted. For the present impurity Cr3+ in tetragonal K2PdCl4 lattice, the electric fields from the rest ions of the crystal could be anisotropic and bring forward some influences on the total crystal fields acting upon Cr3+. To some extent, the opposite contributions to the crystal fields arising from the relevant ions (e.g., Pd2+, Cl-; K+, Cl-; etc.) may partially cancel each other due to opposite charges and lead to insignificant effects on the total crystal fields of Cr3+ because of their successively farther distances
5. Conclusions
The SHPs and local structure for Cr3+ in K2PdCl4 are theoretically analyzed from the perturbation calculations for an orthorhombically distorted 3d3 cluster. The impurity Cr3+ is found not to occupy the host Pd2+ site but to locate at the octahedral K+ site, associated with the relative axial compression ratio

