











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
It is estimated that only 10% of the world aquaculture production is associated with genetic improvement programs; thus, there exists a large potential for increasing production by this approach (Gjedrem et al. 2012). The whiteleg shrimp, Litopenaeus vannamei, is one of the species for which there is growing effort to implement genetic selection programs (Andriantahina et al. 2013). This species is the most important worldwide in shrimp farming, contributing more than 71% of the total production (FAO 2014).
Pedigree tracking is necessary in genetic breeding programs to estimate genetic parameters, including heritability, genotype-by-environment (G × E) interactions, and inbreeding (Gjedrem 2010). The determination of parentage relation- ships among broodstock is also useful when designing breeding plans in order to minimize loss of genetic diversity and inbreeding and maximize productivity (Gjedrem 2010).
Pedigree traceability in genetic breeding programs is usually done by family-based grow-out and/or physical tagging with colored elastomers. This strategy has limitations for individual identification and parentage assignment; hence, genetic tagging has been proposed (Gjedrem 2010). Yue and Xia (2014) summarize several practical considerations for this type of analysis focusing on microsatellites and single nucleotide polymorphisms (SNPs).
The feasibility of microsatellites in parentage assessment has been studied in several shrimp species, such as Marsupenaeus japonicus (Jerry et al. 2004), Penaeus monodon (Jerry et al. 2006), and Fenneropenaeus chinensis (Dong et al. 2006). However, it has been reported that the putative parents of more than 10% of the individuals are usually not accurately assigned (Jerry et al. 2004, Dong et al. 2006, Jerry et al. 2006). This non-assignment proportion has also been reported for fish (e.g., Perez-Enriquez et al. 1999, Christie et al. 2010).
Next-generation sequencing (high-throughput sequencing) has opened the possibility of using SNPs to assess parentage. Lapègue et al. (2014) used SNP markers for pedigree identification of 2 species of cultivated oysters, and suggested using a panel of at least 150 SNPs for a success rate over 95%. For the giant tiger shrimp, P. monodon, Sellars et al. (2014) obtained adequate parentage assignments with panels of 56 and 63 SNPs. In salmon, a 60-loci SNP panel is feasible to genetically differentiate between wild and cultivated stocks (Karlsson et al. 2011).
In the whiteleg shrimp, L. vannamei, genetic markers have mostly been used to characterize genetic diversity and composition of wild and cultured stocks (Cruz et al. 2004, Freitas and Galetti 2005, Perez-Enriquez et al. 2009, Vela- Avitúa et al. 2013). A few studies have focused on individual identification or parentage assignment of breeding stocks (Perez-Enriquez R, unpublished data; Perez F, pers. comm.). The objective of this study was to compare the performance of 2 genetic marker panels (microsatellites and SNPs) for pedigree traceability of a cultivated stock of whiteleg shrimp.
MATERIALS AND METHODS
Sampling
Broodstock from the fifth generation of a commercial breeding program (Acuacultura Mahr hatchery) were selected to obtain 81 full-sib families by artificial insemination. A homogenous number of nauplii per family were placed in a single 20-t tank; larval and juvenile rearing was done following the hatchery's standard procedures until the juveniles weighed 20 g, when a random sample (192 individuals) was collected. Pleopods from broodstock (n = 162) and progeny were preserved in 80% ethanol.
Genotyping
DNA from pleopods was obtained with the Wizard SV 96 Genomic DNA Purification System (A2371, Promega). Microsatellite and SNP genetic profiles were obtained for each individual. A set of 5 published microsatellite loci was used: Pvan1758 and Pvan1815 (Cruz et al. 2002), Lvan05 (Perez-Enriquez et al. 2009), and TumxLv8.256 and TumxLv10.312 (Meehan et al. 2003). Microsatellites Pvan1758, Lvan05, and TumxLv8.26 were amplified by PCR in 11-µL reactions (1 µL DNA at 10-95 ng µL-1, 1× PCR buffer, 3.5 mM MgCl2, 0.25 mM dNTPs, 0.39 µM each of the forward and reverse primers, and 0.025 U µL-1 Taq polymerase) under the authors' reported conditions. For Pvan1815 locus, a touchdown protocol was used (95 ºC for 3 min, 30 cycles at 94 ºC for 45 s, annealing temperature for 45 s [starting at 60 ºC and decreasing each cycle 0.3 ºC down to 51 ºC], 65 ºC for 1 min, and final extension at 65 ºC for 20 min). TumxLv10.312 locus was run using the modified protocol proposed by Yoshida and Awaji (2000) (94 ºC for 2 min; 5 cycles at 94 ºC for 5 s, 60 ºC for 1 min, and 72 ºC for 1 min; 20 cycles at 94 ºC for 1 s, 60 ºC for 1 min, and 72 ºC for 1 min; and final extension at 72 ºC for 10 min). An additional set of 5 loci (Livan04, Livan13, Livan44, Livan51, and Livan60; Table 1) was used. They contained 1.5 mM MgCl2 and the same amount of the other components described above was used with the following amplification conditions: 95 ºC for 3 min; 30 cycles at 94 ºC for 35 s, 57 ºC for 40 s, and 72 ºC for 40 s; and final extension at 72 ºC for 5 min. PCR products were analyzed in an automatic sequencer (ABI3130, Applied Biosystems). Genotypes were read with the software GeneMapper 4.0.

SNP genotyping with a 76-loci panel was performed at the Center for Aquaculture Technologies (San Diego, California, http://aquatechcenter.com/) using allele-specific PCR assays on an EP1 instrument platform (Fluidigm Corporation, San Francisco, CA). Genotypes were analyzed in an Excel file.
Pedigree analysis
Genetic diversity (number of alleles per locus, observed and expected heterozygosity) was estimated for each group (broodstock and progeny) from both marker panels (micro- satellites and SNPs). The probability of identity (likelihood that 2 individuals have exactly the same multilocus genotype) and the combined probability of non-exclusion (likelihood of not excluding a non-true parent) were calculated using Cervus 3.0.3 (Kalinowski et al. 2007). The presence of null alleles and other genotyping artifacts in microsatellites was analyzed with the Microchecker software (Van Oosterhout et al. 2004).
Both panels were used to perform paternity tests to determine the pedigree (family of origin) of each progeny. The analysis was run only for progeny and broodstock with at least 80% of the multilocus genotype complete (i.e., 8 micro- satellite loci or 60 SNP loci).
The parentage assignment tests were done by direct and probabilistic exclusion methods. Direct exclusion, which implies the exclusion of potential breeders with more than 2 mismatching loci, was done with Vitassign software (Vandeputte et al. 2006). This program also performs a simulation to determine the assignment probability from the percentage of unique assignments. Cervus 3.0.3 software (Kalinowski et al. 2007) was used for probabilistic assignment tests. This program gives a minimum assignment value (named LOD) to the pair with the highest likelihood of being the true couple, as a function of the proportion of sampled parents (0.90), genotyping error (0.01), minimum number of genotyped loci, and confidence level (95%). Simulation and real data assignments were compared between microsatellites and SNP markers.
Rarefaction curves were made to estimate the minimum number of SNP loci needed to obtain more than 98% of correct assignments. SNP loci were classified into 2 groups: those with minimum allele frequency (MAF) ≥ 0.3 (both alleles had a frequency between 0.3 and 0.7) and those with MAF < 0.3 (one of the alleles had a frequency of less than 0.3). Four rarefaction curves were drawn based on the proportion (20%, 40%, 50%, and 60%) of loci with MAF ≥ 0.3. Paternity analyses were done with Vitassign software (allowing 2 mismatches) and Cervus software (1.0 proportion of sampled breeders, 0.001 genotyping error, 95% confidence level). The curves were built by obtaining the assignments with 10, 20, 30, 40, 50, 60, and 70 loci, randomly selected from the original set of 76 loci. Only the correctly-assigned progeny from the pedigree test were used for the analyses.
RESULTS
Of the 192 individuals analyzed, complete multilocus genotypes were obtained for 168 using microsatellites (87.5%) and for 186 using SNPs (96.9%). Individuals with incomplete genotypes were not used for the parentage assignment tests.
Higher genetic diversity values (number of alleles per locus, heterozygosity) were obtained with microsatellites than with SNPs; however, the non-exclusion probability was 3 orders of magnitude higher (i.e., a lower probability) with SNPs (Table 2). This characteristic led to a higher percentage of paternity assignment using the SNP panel with both exclusion methods (direct and probabilistic) and with both simulated and real data (Table 2). The use of the 76-loci SNP panel resulted in 94-96% progeny assignment to a unique parental couple, much higher than with microsatellites. The other 4-6% does not correspond to progeny with multiple potential parents, but to a lack of assignment probably due to individuals that originated in a different spawning lot. Null alleles were detected in 3 microsatellite loci (Lvan05, Livan13, and Livan60), which may explain the reduced assignment with these markers.
Table 2 Mean values of genetic diversity (standard deviation in parentheses), probability of identity, combined non-exclusion probability for both parents, and percentage of parentage assignments of progeny (simulated and real data) obtained with microsatellite and SNP panels by direct exclusion and likelihood.
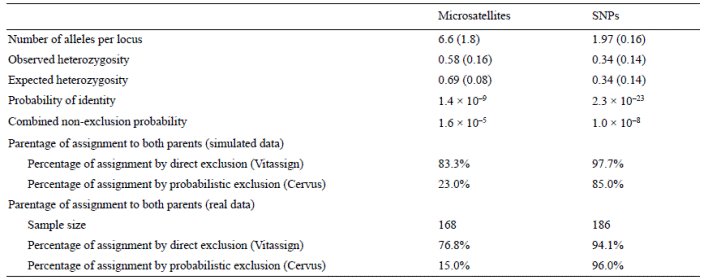
The rarefaction curves indicate that paternity tests by direct exclusion using 50 loci reached 100% assignments when 60% of loci had a MAF ≥ 0.3, and between 98.3% and 99.4% of assignments for MAF ≥ 0.3 proportions of 20%, 40%, and 50% (Fig. 1). The probabilistic assignments at 50 loci reached 100% in 3 of the MAF ≥ 0.3 proportions (40%, 50%, and 60%; Fig. 1).
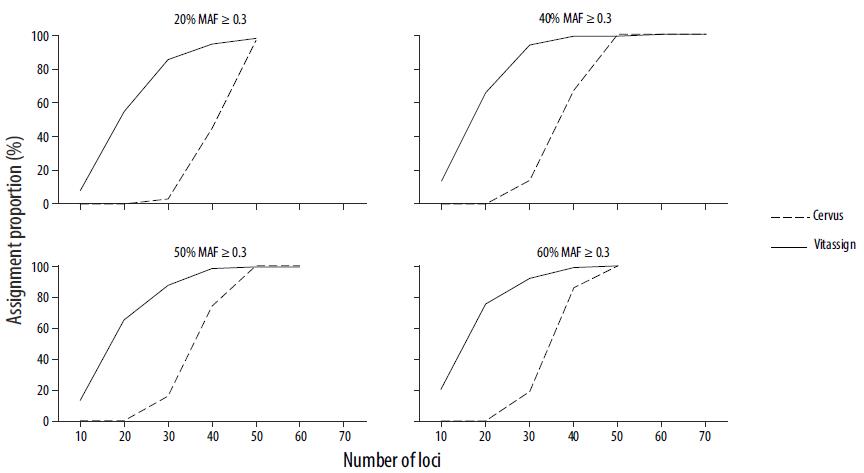
Figure 1 Rarefaction curves of parentage assignment with the SNP panel by direct exclusion (Vitassign software) and probabilistic exclusion (Cervus software), according to the proportion of loci with minimum allele frequency (MAF) ≥ 0.3.
Considering 50 loci as the minimum number for a parent- age assignment analysis, the estimated error (the difference between the assigned parental couple and the true parental couple) showed a decreasing trend as a function of the proportion of loci with MAF ≥ 0.3, the lowest value (2.3%; Fig. 2) being obtained with the 60% proportion.

DISCUSSION
This is the first report to the authors' knowledge of the usefulness of a SNP panel for L. vannamei. It has been demonstrated that a panel of 76 SNPs is suitable to identify the pedigree of cultivated whiteleg shrimp. Despite the lower genetic diversity compared to microsatellites, the certainty of the SNP panel in parentage assignment is higher. The lower resolution of microsatellites than SNPs for this type of application has also been described for tilapia (Trọng et al. 2013) and the giant tiger shrimp, P. monodon (Sellars et al. 2014). Genotyping errors derived from null alleles, allele drop-outs, and mutations (due to a high mutation rate) may explain the lower performance of microsatellites (Ellegren 2000, Pompanon et al. 2005, Trọng et al. 2013, Lapègue et al. 2014). Nevertheless, as previously reported (Trọng et al. 2013), the deletion of loci with high null allele frequency did not contribute to the increase in assignment proportions.
Direct exclusion analysis, allowing a 2.6% of SNP genotyping error (2 mismatches), resulted in >96% of the progeny being assigned, a high assignment certainty. This precisión requires previous knowledge of the mating plan used to obtain the progeny, that is, knowing which male mated with which female. Nevertheless, in those cases where the mating plan is unknown, use of the SNP panel is still feasible because likelihood analysis is capable of identifying the families with an error of less than 3% and 95% confidence. Trọng et al. (2013) reported similar assignment levels with both analytical methods (direct and probabilistic exclusion) for a cultivated tilapia population.
Even though the number of loci of the SNP panel (76) is slightly higher than the number of loci used by Sellars et al. (2014) for P. monodon (53 and 63 SNPs), the results indicate that, when the proportion of loci with MAF > 0.3 in the panel is 60%, 50 loci would be enough for the correct assignment of more than 98% of the progeny. These results contrast with those obtained by Lapègue et al. (2014) with oysters and Trọng et al. (2013) with tilapia who used panels of between 122 and 384 loci. In fact, Lapègue et al. (2014) recommends a minimum of 150 loci with a mean MAF of 0.3 to obtain a robust assignment.
Assignment probability is a function of the population allele frequencies (Kalinowski et al. 2007); therefore, related individuals within the broodstock, which share more alleles than unrelated individuals, will decrease the probability of correct assignment. In this regard, Trọng et al. (2013) obtained a low to moderate precise assignment in tilapia as a result of high genetic similarity among individuals, due to more than 8 generations of genetic selection. In this study, the broodstock belonged to a fifth generation of selection for which inbreeding has been maintained at a low level (1.1-2.4%; Perez-Enriquez et al., unpublished data). Although this low inbreeding seems to have no effect on the assignment precision when using SNPs, some microsatellite loci might have been affected, increasing the frequency of null alleles. Information on broodstock management history is always recommended when genetically tracing pedigree.
The certainty in pedigree assignment using SNP markers opens the possibility for experimental studies in a common environment for estimating heritability, G × E interactions, and broodstock management when physical tagging is not possible or efficient. With the analysis platforms presented here, the use of SNPs in shrimp is suggested as a higher benefit/cost alternative (cost per sample, genotyping rate, analysis time) than microsatellites.

