











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Herbal preparations are mixtures of compounds that individually, or in combination, appear to provide beneficial health effects. Therefore, they are widely used as complementary medicines or nutraceuticals (Izzo 2005). However, several reports have shown adverse drug interactions with medicines from terrestrial plants, such as St. John’s wort, garlic, ginseng, Gingko biloba, soya beans, alfalfa and grape fruit juice (Obach 2000, Zhou et al. 2003, Bailey et al. 2004, Liu et al. 2006, Zhou et al. 2007). Cytochrome P450 enzymes (CYP) and ABC transporters are key mediators of herb-drug interactions (Pal and Mitra 2006, Aszalos 2008). Such interactions could have serious effects on the pharmacokinetics and toxicity of pharmaceuticals.
Thalassia testudinum Banks ex Koenig, an abundant coastal sea grass found in Cuba has been subject to phytochemical and pharmacological studies (Regalado et al. 2012, Rodeiro et al. 2012). The aqueous-ethanol extract obtained from the leaves of this marine plant is rich in polyphenols (Regalado et al. 2009, de la Torre et al. 2012, Regalado et al. 2012). The skin protective activity-guide fractionation of the extract showed thalassiolin B (chrysoeriol 7-b-D-glucopyranosyl-2”-sulphate) as its main active component (Regalado et al. 2009). Further analysis revealed the presence of other phenols like apigenin 7-O-β-D-glucopyranosyl-2”-sulfate (thalassiolin C), chrysoeriol 7-O-β-D-glucopyranoside, apigenin 7-O-β-D-glucopyranoside, dihydroxy-3’,4’-dimethoxyflavone 7-O-β-D-glucopyranoside, luteolin-3’-sulphate, chrysoeriol and apigenin. The pharmacological properties of the extract include anti-inflammatory and antioxidant effects (Nuñez et al. 2006, Rodeiro et al. 2008a, Regalado et al. 2012). Neuroprotective properties have also been reported in rodents, which may represent a therapeutic application of the extract (Garateix et al. 2011, Menendez et al. 2014). Due to these beneficial effects, a food supplement is being developed from the extract.
Previously we evaluated the effects of T. testudinum extract on the CYP enzymes activity in cultured rat hepatocytes and in human liver microsomes (Rodeiro et al. 2008b, Rodeiro et al. 2009). We observed a reduction in the activity of CYP2E1 in rat hepatocytes, while an increase in the activity of CYP2B1 was obtained after 48 hours of exposure. The activity of the rest of the tested P450 cytochromes remained unchanged. The extract markedly reduced human liver microsome CYP1A2 and CYP3A4 activities (Rodeiro et al. 2009). In addition, significant reduction in CYP1A2, CYP3A4 and CYP2D6 activities were observed in human hepatocytes exposed to the extract for 48 hours (5-100 μg/mL). Quantification of specific mRNAs confirmed these findings for CYP3A4. In contrast, increased CYP1A1 and CYP1A2 mRNAs were found (Rodeiro et al. 2012). These results suggest that activity and expression of major CYP isoenzymes are modulated by the extract and that herb-drug interactions could arise if this herb is used with conventional medicines. Moreover, the extract may interfere with bioconversion of environmental carcinogens. As the extract modulates in vitro the function of these phase I enzymes, it should be examined the potential for in vivo interactions of the product. Analysis should examine alterations of the absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of other xenobiotics used concomitantly with the extract.
Here, we study the effects of oral doses of the extract (20 to 400 mg/kg) on the activity of 1A, 2B, 2E and 3A liver CYP subfamilies in male Wistar rats. We further examined the effects on protein and mRNA levels on those isoforms whose activities were modulated by the T. testudinum extract. Finally, we looked for confirmation of observed effects by evaluating the interaction of the extract with the biotransformation of theophylline and benzo[a]pyrene.
MATERIALS AND METHODS
Chemicals
Ethoxyresorufin, methoxyresorufin, benzoxylresorufin, pentoxyresorufin, dimethyl sulfoxide (DMSO), 4-nitrophenol, 4-nitrocatechol, benzo[a]pyrene and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) were purchased from Sigma Chemical Co. (St Louis, MO). Rabbit polyclonal anti-rat CYP1A1/2 (Chemicon International Inc.), mouse polyclonal anti-rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore), chemicals for electrophoresis and nitrocellulose membranes were purchased from Bio-Rad Laboratories (Richmond, CA). Trizol® reagent, Moloney murine leukemia virus reverse transcriptase (M-MLV RT) and oligodeoxythymidylic acid (oligo dT) primers (Invitrogen), Taq Man® universal polymerase chain reaction (PCR) master mix, unlabeled PCR primers and TaqMan® minor groove binder (MGB) probes were purchased from Applied Biosystems (Foster City, CA, USA).
Plant material
The extract was provided by the chemistry department of Marine Bioproducts Center (CEBIMAR) (batch 0902001) as a fine, homogeneous green powder with the typical odor (CEBIMAR, Analytical Service Results Report). Thalassia testudinum Banks and Soland ex. Koenig was collected in April 2009 from “Guanabo” Beach (22º 05’ 45’’N, 82º 27’ 15’’ W) and was identified by Dr. Areces J.A. (Institute of Oceanology, La Habana, Cuba). A voucher sample (No. IdO 039) was deposited in the herbarium of the Cuban National Aquarium. Dried and ground leaves of T. testudinum (2.3 kg) were extracted with 30 L of EtOH/H2O (50:50) (3 × 10 L) at room temperature. The combined solutions were filtered, concentrated under reduced pressure and dried by sprinkler to yield 170 g of extract. Characterization was performed qualitatively and quantitatively by two standard phytochemical screening tests, as described by Regalado et al. (2012). The results were: total phenolic content (29.5 ± 1.2 %), flavonoids (4.6 ± 0.2 %), proanthocyanidins (21.0 ± 2.3 %), polysaccharides (5.8 ± 1.6 %), lipids (0.59 ± 0.01 %), soluble proteins (16.2 ± 0.7 %) and chlorophylls a and b (3.43 and 1.44 μg/mL, respectively) (Regalado et al. 2012). Thalassiolin B (5.8 ± 0.9 %) was quantified as reported previously (Garateix et al. 2011). Details are available as supplementary data on Regalado et al. (2012). For the experiments, the extract was dissolved in distilled water (vehicle).
Effect of the extract on CYP activity, protein and mRNA levels
Animals
Male Wistar rats (180-200 g) were obtained from the Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México (Biomedical Research Institute, National Autonomous University of Mexico (UNAM, Mexico City, Mexico). Animals were acclimatized under constant conditions (temperature 20 ± 2 ºC, humidity 40-60 %, and 12 h light/dark cycle) for one week. They were fed with standard commercial rat diet and water ad libitum. The experiment was conducted in accordance with the ethical guidelines for investigations with laboratory animals and it was approved by the ethical committee for animals.
Treatments
The study was conducted in four experimental groups (five animals/group). Animals were administered with oral doses of the extract (20, 200 or 400 mg/kg of bodyweight) for 10 days. The control group received distilled water (vehicle). Animals were starved for 12 h before sacrifice to prevent severe hepatic metabolism variations. They were weighed before being sacrificed by cervical dislocation. Livers were rapidly removed, weighed, washed in a 0.15 M KCl solution and stored at -80 ºC until RNA extraction or microsomal preparation.
Preparation of liver S9 and microsomal fractions
Liver S9 fractions were prepared as described by Maron and Ames (1983). Livers were removed, homogenized in KCl 0.15 M and centrifuged at 9000 g for 10 min. The supernatant was stored at -80 ºC. A portion of the supernatant was centrifuged at 105 000 g for 60 min at 4 ºC. The pellet was resuspended in phosphate buffer (67.5 mM K2HPO4, 32.5 mM KH2PO4, pH 7.4) and centrifuged again. Microsomes were stored at -80 ºC in buffer with 1 mM dithiothreitol, 1 mM EDTA and 20 % glycerol. The protein content was determined by the Bradford method (Bradford 1976).
CYP1A1 and CYP1A2 activities
CYP1A activity was determined by measuring the formation of resorufin by the O-dealkylation of 7-ethoxyresorufin for CYP1A1 and of 7-methoxyresorufin for CYP1A2. Measurements were performed by fluorescence spectroscopy according to Burke et al. (1994). Buffer (50 mM Tris-HCl, 25 mM MgCl2, pH 7.6), substrate and NADPH were incubated at 37 ºC for 3 min in a cuvette, and the reactions were initiated by adding microsomal protein (200 μg). Reactions were monitored for 3 min by recording fluorescence every 15 s. Activities were calculated from a standard resorufin curve (5-500 pmol/mL). Excitation and emission wavelengths were set at 530 and 590 nm, respectively.
CYP2B1 and CYP2B2 activities
CYP2B activities were determined by measuring the formation of resorufin by O-dealkylation of 7-penthoxyresorufin (CYP2B1) and 7-benzoxyresorufin (CYP2B2) according to Burke et al. (1994). The reaction was conducted as described for CYP1A1/2 activities.
CYP2E1 activity
CYP2E1 activity was monitored by measuring through spectrophotometry, the biotransformation of 4-nitrophenol hydroxylation to give 4-nitrocatechol (Reinke and Moyer 1985). Microsomal protein (1 mg) was suspended in buffer (50 mM Tris-HCl, 25 mM MgCl2, pH 7.6) to a final volume of 2 mL and it was incubated with 4-nitrophenol (0.25 mM) for 5 min at 37 ºC. The reaction was initiated by adding 50 mM NADPH and the incubation continued for 10 min. The reaction was stopped by adding 0.5 mL of 0.6 N perchloric acid followed by centrifugation. The supernatant (1 mL) was mixed with 0.1 mL of 10 N NaOH and 4-nitrocatechol was measured at 510 nm. Activities were calculated from a standard curve of 4-nitrocatechol (5-50 nmol/mL).
CYP3A activity
Erythromycin N-demethylation was assessed by measuring the production of formaldehyde (HCHO) in a spectrophotometer as previously described (Wrighton et al. 1985). The incubation mixture (1 mL final volume) included 0.1 mL of MgCl2 (150 mM), 0.1 mL of erythromycin (100 mM), 0.1 mL of microsomal protein and phosphate buffer (50 mM, pH 7.25). The mixture was pre-incubated at 37 ºC for 3 min and the reaction was initiated with 0.1 mL of NADPH (10 mM in 1 % NaHCO3). After 10 min, the reaction was terminated by the addition of 0.5 mL of 12.5 % trichloroacetic acid (TCA). After centrifugation, 1 mL of supernatant was mixed with 1 mL of Nash reagent, heated at 50 ºC for 30 min and the absorbance was measured at 412 nm. A standard HCHO solution (1 μM/mL) was used for the calibration curve.
Western blot analysis
Aliquots were taken from the microsomal fractions of each animal in all groups. Tissues from the same group were mixed in order to produce a pool for each group and 10 μg of protein was assayed. Proteins were separated in 7.5 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 mm nitrocellulose sheets overnight (Towbin et al. 1979). Nitrocellulose membranes were submerged for 1 h in 5 % non-fat dry milk diluted in phosphate buffer saline at 4 ºC (Guengerich et al. 1982). After 10 min wash with phosphate-buffered saline (PBS) and 0.3 % Tween, membranes were incubated in the presence of rat primary antibody (anti-CYP1A1/2). CYP protein levels were normalized with Ponceau staining as the loading control. Proteins were revealed with luminol. Relative increases in band intensity over controls for CYP isoforms were determined with a computer program (1D Kodak 3.6.3).
RNA isolation and expression analysis. RNA was isolated from the liver using TRIzol® Reagent according to the manufacturer’s instructions. RNA concentration and purity were determined by absorbance at 260/280 nm, and RNA integrity was tested by electrophoresis on a 1 % denaturing agarose gel. Single-stranded cDNA was synthesized from 1 μg of total RNA by a reverse-transcription reaction with 500 units of M-MVL RT and oligo dT primers. Quantitative PCR was performed using an ABI PRISM 7500 Sequence Detection System (Applied Biosystems). Taq-Man® Gene expression primers (Applied Biosystems) were used, primer Rn00487218_m1 for CYP1A1, Rn 00561082_m1 for CYP1A2 and Rn 99999916_s1 for GAPDH. Quantitative gene expression was calculated from the cycle threshold value of each sample in the linear portion of the curve. The 2-ΔΔCT relative quantification method was used (Livak and Schmittgen 2001). All samples were assayed in triplicate; the mean values of each gene were normalized to the mean for the housekeeping gene, GAPDH.
Benzo[a]pyrene biotransformation by hepatic S9
The Salmonella mutagenicity plate incorporation test was carried out according to Maron and Ames (1983) to confirm the CYP1A1 results. Biotransformation of benzo[a]pyrene (10 μg/plate) was monitored using the TA98 Ames strain. For each test, 2 mL of top agar (0.6 % agar, 0.5 % NaCl, 0.5 mM biotin and 0.05 mM L-histidine) was mixed with 0.1 mL of the overnight culture (approximately 108 cells) and 0.5 mL of S9 fraction. S9 were obtained from animals treated with the extract (20, 200 and 400 mg/kg) or vehicle (control group). All plates were incubated at 37 ºC for 48 h, and revertant colonies were counted.
Pharmacokinetics of theophylline in rats treated with the extract
Animals
Male Wistar rats (180-200 g) were obtained from Centro para la Producción de Animales de Laboratorio, Havana, Cuba (CENPALAB). Animals were acclimatized under constant conditions (temperature 20 ± 2 ºC, humidity 40-60 %, and 12 h light/dark cycle) for one week. They were fed with standard commercial rat diet and water ad libitum. The experiment was conducted in accordance with the ethical guidelines for investigations with laboratory animals and it was approved by the CENPALAB ethical committee for animals.
Treatments
Rats were divided into four groups; they received daily oral doses of the extract (20, 200 or 400 mg/kg) or distilled water for 10 days. The rats were anaesthetized with 25 mg/kg of sodium pentobarbital, and they were cannulated in the right jugular vein for blood sampling. Eighteen hours later, 10 mg/kg of theophylline was orally administered. Blood samples were taken at 0, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after theophylline administration. The plasma samples were separated by centrifugation at 2000 g for 10 min and stored at -20 ºC until analysis.
Analytical methods
A 50 μL aliquot of caffeine (20 μg/mL in mobile phase) was added as internal standard (IS) to a 100 μL plasma sample in a screw-cap glass tube. The mixture was shaken vigorously for 30 s and extracted with 3 mL of diethyl ether-dichloromethane (3:2, v/v). Samples were mixed vigorously for 2 min and later they received centrifugation at 2500 g for 10 min, the organic layer was separated and evaporated to dryness at 40 ºC under a gentle stream of nitrogen. The residue was reconstituted in a 100 μL mobile phase followed by vigorous shaking. A 20 μL aliquot of the supernatant was subjected to high performance liquid chromatography (HPLC).
HPLC separation of theophylline was carried out at room temperature on a reverse phase phenomenex aqua C18 column (250 × 4.60 mm, 5 μm particle size). The mobile phase (methanol/1 % formic acid 28:72, v/v) was delivered at 1.0 mL/min. Calibration curves were linear over the range of 0.1-25.0 μg/mL. The lower limit of quantification was 0.1 μg/mL, with a precision < 7 % and an accuracy of 105 %. Recovery of theophylline ranged from 82 to 88 %, coefficient of variation (CV) less than 8 %. Within-day and between-day CVs for this assay were < 7 %.
Pharmacokinetics
The elimination rate constant (Ke) was calculated by the least squares regression method. The last four points of the plasma concentration-time curve were used. The half-life (t1/2) of the drug was calculated as 0.693/Ke. The area under the plasma concentrationtime curve from t0 to the last measurable plasma concentration (AUClast) was calculated by the linear trapezoidal rule. The AUC from t0 to t∞ (AUCinf) were obtained as AUClast + Ct/Ke. The maximum plasma concentration (Cmax) and the time to reach maximum concentration (Tmax) were determined by visual inspection.
Statistical analysis
Ames test results (revertants/plate) were reported as mean ± standard deviation (SD) and compared by the non-parametric Kolmogorov-Smirnov test using Salanal statistical software. Body weight gain, relative liver weight, enzyme activity, protein and mRNA levels and pharmacokinetic parameters were expressed as the mean ± standard error of mean (SEM). These data were analyzed using Student’s t-test, p values below 0.05 were considered statistically significant.
RESULTS
Effect of the extract on phase I enzyme activity
No significant differences in body weight gain or relative liver weight were observed for the different doses of the extract (data not shown). The effects of oral 20, 200 and 400 mg/kg doses of the extract on ethoxyresorufin O-deethylation (EROD) (CYP1A1), methoxyresorufin O-demethylation (MROD) (CYP1A2), pentoxyresorufin O-dealkylation (PROD) (CYP2B1), benzyloxyresorufin O-dealkylation (BROD) (CYP2B2), 4-nitrophenol hydroxylase (PNPH) (CYP2E1) and erythromycin demethylase (END) (CYP3A) liver activities of male Wistar rats treated for 10 days are shown in figure 1. The extract at 200 mg/kg induced a significant 1.5-fold increase of EROD activity in liver microsomes. Meanwhile, after the addition of 400 mg/kg, a small increase was observed, but it was not significantly different compared to control groups. A low dose of the extract (20 mg/kg) induced an increase in CYP1A2 and CYP2B1 activity (~ 20 %) when compared to control groups, but statistical significance was not attained. A slight, but no significant reduction (~ 20 %) of CYP2B1/2, 2E1 and 3A activity was observed at higher doses.
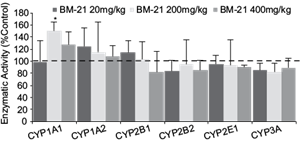
Fig. 1 Effects of the Thalassia testudinum extract on male Wistar rat liver microsome P450 activities. BM21 = Talassia testudinum extract. Cytochrome P450 = CYP1A1, CYP1A2, CYP2B1, CYP2B2, CYP2E1 and CYP3A. The results are expressed as percentage of control group, values represent the mean ± standar deviation of 5 animals/group. * < p 0.05 respect to control group.
Taking into account the increased values of CYP1A1 activity, it was carried out a western blot analysis in liver microsomes from treated rats. Increased significant expression of CYP1A1/2 protein was found at 20 and 200 mg/kg (Fig. 2). As 1A1/2 polyclonal antibody recognized both isoforms, this result may explain why this isoform was induced, thus suggesting that the increased activity of CYP1A1 enzyme was partly due to increased protein expression.
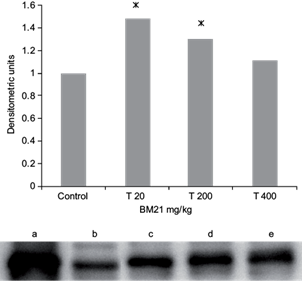
Fig. 2 Effects of the Talassia testudinum extract on male Wistar rat liver CYP1A1/2 protein levels. Values are expressed as the fold change relative to the control group. Lanes: (a) CYP1A1/2 Supersome®, (b) control, (c) T20 mg/kg, (d) T200 mg/kg and (e) T400 mg/kg. Columns represent the band intensity. BM21 = Talassia testudinum extract. Samples from five animals/group were evaluated. * < p 0.05 respect to control group, student t-test. CYP = cytochrome P450, T = treatment
Oral exposure to the extract did not significantly change CYP1A1 and 1A2 mRNA levels relative to control groups, except for a reduction in mRNA level at 20 mg/kg dose for CYP1A1 (Fig. 3). Therefore, increased protein level and activity observed for these isoforms, cannot be explained from variations in the transcription of the associated genes.

Interaction with phase I biotrasformation of benzo[a]pyrene (Ames test)
Increased activity of CYP1A isoforms may produce changes in xenobiotics metabolism. It is well known that benzo[a]pyrene metabolism occurs in liver and it is a substrate for these isoforms. The mutagenicity of benzo[a]pyrene increased in cells incubated with the S9 fractions from rats previously treated with the extract. Significant differences were: 1.8-fold increase for 200 mg/kg and 2.3-fold increase for 400 mg/kg of the extract when compared to the control groups. Interestingly, mutant colonies reduced to 87 at 20 mg/kg doses (Table I), which may be explained as a hormetic response (Calabrese et al. 2011). However, the difference with the control is in the normal range of fluctuation for the Ames test. More experiment should be carried out in order to improve the comprehension of the interaction among the extract and the enzymes of the P450 system.
TABLE I EFFECTS OF Thalassia testudinum EXTRACT ON THE BIOACTIVATION OF BENZO[A]PYRENE USING Salmonella typhimurium TEST (TA98 STRAIN)
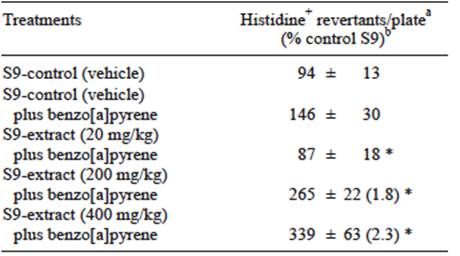
aValues represent the mean ± SD of histidine revertants: two independent experiments were carried out in triplicate. bInside the parenthesis is the ratio of revertants relative to 200 and 400 mg/kg over S9-control group (vehicle) plus benzo[a]pyrene. * p < 0.05 compared to S9 control group (vehicle) plus benzo[a]pyrene treatment, Mann Whitney test. S9 = rat liver microsomes
Effects on the pharmacokinetics of theophylline in rats
Theophylline plasma concentrations increased significantly during the first 2 h after receiving the drug in male Wistar rats previously treated for 10 days with oral doses of the extract (Fig. 4). The extract duplicated the Cmax value relative to the control group. Clearance of theophylline was also increased, affecting the mean elimination rate constant (Ke) and half-life (t1/2). Ke showed a 2-fold increase while t1/2 was reduced twice regarding control groups (Table II). No significant differences were observed among groups for any of the recorded parameters.
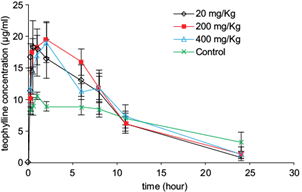
Fig. 4 Mean theophylline plasma concentration vs. time curves in rats treated with the Talassia testudinum extract. Each point indicates the mean ± standard error of mean of theophylline plasma concentrations
TABLE II EFFECTS OF Thalassia testudinum EXTRACT ON THE METABOLISM OF THE THEOPHYLLINE IN MALE WISTAR RATS
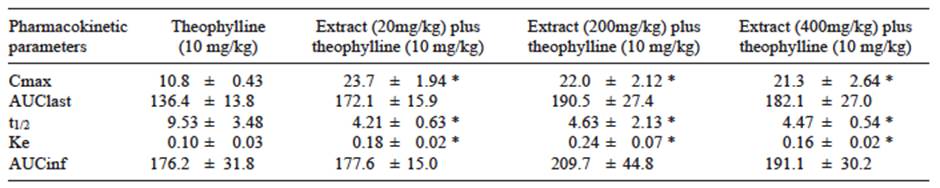
Cmax = maximum plasma concentration, AUClast = area under the plasma concentration - time curve from time cero to the last measurable plasma concentration, t1/2 = half-life time, Ke = elimination rate constant, AUCinf = area under the whole curve. Data are expressed as the mean ± standard error of mean (n = 6), * p < 0.05 vs. control group (student t-test).
DISCUSSION
Biotransformation is the cellular process of modifying xenobiotics they enter into contact and whose ultimate goal is to facilitate their elimination. To achieve this, lipophillic compounds, which otherwise would accumulate in intracellular lipids, become more hydrophilic under the activity of a set of broad-specificity enzymes. These enzymes are capable of either introducing new functional groups (phase I reactions) or conjugating with hydrophilic endogenous molecules to increase water solubility (phase II reactions). The formation of water-soluble metabolites not only enhances drug elimination, but may lead to molecules which are pharmacologically inactive and relatively non-toxic, although in many cases toxic metabolites can be formed (Zhou et al. 2007).
Induction and/or inhibition of phase I and II metabolism are the main factors responsible for drug interactions with other xenobiotics. A characteristic of many P450 enzymes is their capacity to be induced by xenobiotics. Upon repeated administration, certain drugs can increase their own metabolism (or the one of other agents) by altering the expression or activity of P450s and other drug metabolizing enzymes. Moreover, as xenobiotics are metabolized by a limited number of enzymes, they can compete among themselves as substrates, inhibitors or inducers for P450 enzymes (Xie et al. 2016). Hence, the presence of one chemical can interfere with the metabolism of another. Induction of a metabolically relevant P450 by a particular compound can result in decreasing the plasma/tissue concentration of other co-administered drugs. Changes in pharmacokinetic properties often produce inadequate or variable clinical response to the drug that can compromise its therapeutic usage (de Sande 2011). Given the importance of P450 phenotype in pharmacokinetics, pharmacodynamics and the potential toxicity of a given drug, the identification of potential factors altering the enzyme expression and/or function is relevant. Such phenomena are at the root of potential drug-drug interactions (de Sande 2011).
Consequently, interactions between natural products and pharmaceuticals can occur, leading to undesired effects. These, include impaired bioavailability of drugs with narrow therapeutic indices, altered plasma/tissue levels, and enhanced bioactivation of drugs to reactive intermediates or toxic metabolites (Hedrich et al. 2016).
Our previous in vitro results demonstrate that the extract from the marine plant T. testudinum can modulate CYP activities in cultured hepatocytes and human liver microsomes (Rodeiro et al. 2008b, Rodeiro et al. 2009, Rodeiro et al. 2012). The present work provided the first in vivo evidence regarding the effects of this natural extract on the main CYP isoforms involved in the metabolism of several xenobiotics. Except for CYP2B1, no increase in CYP activity is previously observed in vitro. In contrast, this study showed that CYP1A1 activity increased after 10 days exposure to oral doses of the extract at 200 mg/kg (Fig. 1). Thus, the extract induced effects on CYP1A1 in vivo that are not observed in cultured rat hepatocytes, suggesting differences in extract-mediated metabolism in vitro and in vivo under our experimental conditions. These results suggested that the absorption of the extract components or the gastrointestinal metabolism may play an important role in the modulation capacity of the extract over the CYP enzymes in rats.
In terms of molecular mechanism, changes in the activity of drug-metabolizing enzymes can be divided into transcriptional regulation or direct effects on protein activity. Our immunoblot analyses revealed that rat CYP1A1/2 protein levels also increased after oral administration of the extract, consistent with the elevated activity of these isoforms after treatment. These results suggested that the extract components may stabilize CYP1A1/2 protein because no changes in mRNA levels were evident (Fig. 3). However, a direct effect on the catalytic activity of CYP1A1 cannot be discounted.
CYP1A2 is expressed mainly in the liver (Rendic and Di Carlo 1997). This constitutes approximately 10-15 % of the total CYP content in the human liver. CYP1A2 metabolizes important drugs, such as phenacetin, theophylline, caffeine, imipramine, propranolol, clozapine, tacrine (Brosen 1995, Cupp and Tracy 1998), and it also activates some pro-carcinogens (Smith et al. 1996). Theophylline is predominantly metabolized by CYP1A2, while CYP2E1 and CYP3A4 are thought to have only minor roles (Gu et al. 1992, Bachmann et al. 1993). Pharmacokinetic analysis of serum theophylline concentrations was performed to assess the pharmacokinetic interactions of theophylline with the extract. The results demonstrated that oral administration of the extract to rats stimulated metabolite formation. The extract did not significantly change the area under the plasma concentration (AUC) values for theophylline, but clearance was significantly increased. The t1/2 of elimination was reduced, while the elimination rate constant was increased (Table II). These pharmacokinetic changes suggested that ingestion of the extract facilitated the excretion of theophylline in rats. This, in turn, revealed a possible risk of interactions if the extract is consumed with theophylline. In summary, our results demonstrated that ingestion of the extract elevates CYP1A activity.
The liver plays a pivotal role in the activation and elimination of xenobiotic toxins. Biotransformation enzymes, such as CYP, are essential to convert lipophilic xenobiotics into more hydrophilic, watersoluble metabolites (Gomez-Lechón et al. 1997). At the same time, induction of phase I enzymes is considered a potential risk factor for cancer due to the activation of pro-carcinogens to carcinogens (Androutsopoulos et al. 2009).
The effect of the extract on the mutagenicity of benzo[a]pyrene was assessed by the Ames test. TA98 cells were incubated in the presence or absence of benzo[a]pyrene, which is activated by CYP1A1 to give a human carcinogen. In this assay, we used the S9 mixture obtained from animals treated orally with extract or from control groups. The S9 fraction prepared from rats treated with the highest doses of the extract did activate the enzymes which transform benzo[a]pyrene significantly more efficiently than the one prepared from control groups (Table I), but it has the opposite effect at 20 mg/kg dose. This evidence suggested that even though the extract interferes with CYP1A1 activity, the nature of such interaction is a complex one, therefore more evidence should be collected in order to fully understand its nature. We suggest that the extract may facilitate the bioactivation of xenobiotics to highly reactive metabolites capable of interacting with important macromolecules such as DNA and proteins.
CONCLUSION
In summary, our present results indicate that the extract modified the activities and expression of CYP1A, enzymes responsible for the biotransformation of agents such as theophylline and benzo[a]pyrene. Significant in vivo metabolism-based pharmacokinetic interactions between the extract and theophylline occurred in rats. Although direct extrapolation from rats to humans is difficult, our results reveal potential drug interactions with the extract. Herbdrug interaction may occur when patients use the extract in combination with CYP1A substrates.

