











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The class I myosins are a family of actin-dependent molecular motors involved in different functions1. Humans possess eight genes that code for these proteins (Myo1a-Myo1h) and are subdivided into short-tailed (Myo1a,b,c,d,g,h) and long-tailed (Myo1e,f) myosins2. Class I myosins have attracted interest because some function as tumor suppressors, and others are overexpressed in different types of cancer. We recently identified Myo1g as a potential diagnostic biomarker for childhood acute lymphoblastic leukemia (ALL). Myo1g is expressed exclusively in hematopoietic cells, with a high expression in B and T lymphocytes3-6. Myo1g generates the membrane tension required for T and B cell migration, endocytosis, exocytosis, and phagosome closure. We found that Myo1g is overexpressed at different stages of the disease, mainly at diagnosis, and proposed that Myo1g could serve as a marker of high risk and early relapse to contribute to improved patient survival7.
ALL is the most common pediatric malignancy and accounts for at least 25% of childhood cancer8. Although relapse of ALL is frequent, immunophenotype change (lineage switch) rarely occurs at relapse9,10. Lineage switch accounts for 6-9% of relapse cases and is more frequent in pediatric patients11. Most reports of lineage switch arise from ALL to acute myeloid leukemia (AML) conversions12-14. However, the underlying mechanisms are poorly understood. Moreover, the prognosis for these patients is variable, and there is no standard treatment15.
ALL immunotherapy with blinatumomab has been beneficial in treating patients with refractory or relapsed forms of the disease. Up to 20% of post-blinatumomab relapses are characterized by loss of CD19 from leukemic cells. Some CD19-negative relapses are accompanied by loss of all B-lineage antigens and acquisition of a distinct myeloid immunophenotype. The KMT2A/AFF1 rearrangement in B-ALL is an independent poor prognostic factor associated with a higher rate of treatment failure and an increased risk of linage switch under therapy16,17.
Diagnosis can be difficult and delayed in the risk staging for atypical or early-stage cases according to the institutional protocol. Since we recently reported that Myo1g is a risk biomarker7, we decided to follow this case to determine if Myo1g could aid in the diagnosis or follow the etiology of the lineage switch.
Here, we present the first case documented by immunofluorescence (IF) and qRT-PCR of the role of Myo1g in the clinical follow-up of ALL with lineage switch to myeloid phenotype.
Clinical case
We describe the case of a 1-year-old Mexican female patient admitted for hepatomegaly. Her parents were not consanguineous, and she was the youngest of three siblings from a humble community. She was delivered by cesarian section at nine months due to cephalopelvic disproportion (CPD) without complications. She weighed 3,700 kg and was hospitalized for five days due to hyperbilirubinemia. The complete neonatal screening was normal, but the vaccination schedule was incomplete. Physical examination at hospital admission showed left preaxial polydactyly, developmental delay (sitting up at 11 months), and hepatomegaly. No other relevant signs were observed.
Hepatic infiltration of B-cell precursors
In November 2013, different laboratory tests were performed, including blood biometry and serological tests for TORCH, viral hepatitis, hepatovirus, and Epstein-Barr virus, showing no alterations. The results for Gaucher and Niemann-Pick diseases were negative.
Two days later, she developed a fever and was diagnosed with a fever of unknown origin. Thick blood drop study, BM culture, and complement were also normal. Imaging studies (computerized axial tomography) showed cervical and inguinal lymphadenopathy and hepatosplenomegaly. A liver biopsy showed infiltration by B-cell precursors, CD10 +, CD19 +, CD20 +, and TdT + neoplastic cells (Figure 1). Blood biometry was performed again. The white blood cell count (WBC) was 20,400/μL with 29% neutrophils, 50% lymphocytes, 4% monocytes, 0% eosinophils, and 6% blasts. Hemoglobin was 10.7 g/dL, and platelet count was 77,000/μL (Table 1).
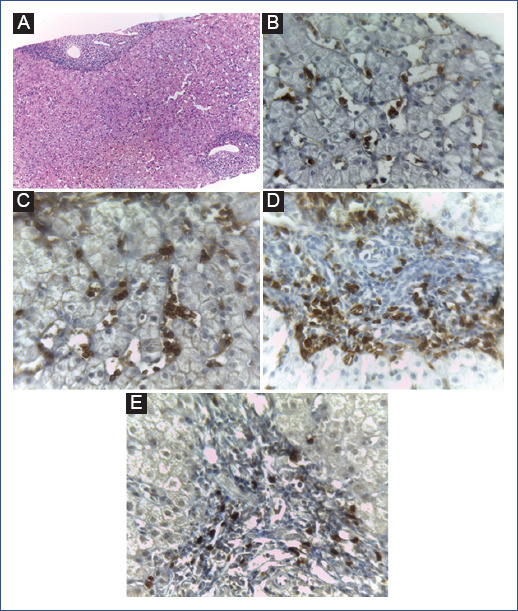
Figure 1 B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in liver biopsy. A: hematoxylin and eosin staining (40x). Immunohistochemical staining showed that the neoplastic cells were positive for B: CD10 (200×), C: CD19 (200×), D: CD20 (200×), and E: TdT (200x).
Table 1 Laboratory, immunophenotypic, and genetic findings
| CBCC | ALL diagnosis | First relapse | Second relapse |
|---|---|---|---|
| WBC | 20,400/μL | 31,800/μL | 43,000/μL |
| Blasts | 6% | 15% | 18% |
| Segmented | 29% | 21% | 32% |
| Eosinophils | 0% | 0% | 3% |
| Basophils | 0% | 0% | 5% |
| Monocytes | 4% | 9% | 35% |
| Lymphocytes | 50% | 70% | 25% |
| RBC | 3.80 x106/μL | 3.65 x106/μL | 2 x106/μL |
| Hemoglobin | 10.70 g/L | 10.40 g/L | 6.5 g/L |
| PLT | 77,000/μL | 95,000/μL | 19,000/μL |
| Bone marrow | |||
| Blasts | 14.5% | 37% | 30% |
| CD19 | + | + | - |
| CD20 | - | - | - |
| CD22 | + | + | - |
| CD79a | + | + | - |
| sKAPPA | + | - | - |
| sLAMBDA | + | - | - |
| CD13 | - | - | + |
| CD14 | - | - | - |
| CD15 | - | + | - |
| CD33 | - | + | + |
| CD2 | - | - | - |
| CD3 | - | - | - |
| CD5 | - | - | - |
| CD7 | - | - | - |
| CD10 | + | - | - |
| CD34 | - | + | + |
| CD41 | - | - | - |
| CD45 | + | + | + |
| CD117 | - | - | + |
| HLA-DR | - | - | + |
| Glycophorin | - | - | - |
| TdT | - | - | - |
| MPO | + | - | + |
| TP | - | - | WND |
+: positive; −: negative.
ALL: acute lymphocytic leukemia; CBCC: complete blood cell count; MPO: myeloperoxidase; PLT: platelets; RBC: red blood cells; TP: translocation panel; WBC: white blood cells; WND: was not done.
Risk staging
According to the institutional protocol and the 2008 WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, the patient's conditions indicated a low-risk situation with a good prognosis due to age, absence of hyperleukocytosis and not otherwise specified (NOS) genetic abnormalities. The session with the hematology, oncology, and pathology departments provided the diagnosis of BCP-ALL of hepatic origin. The patient started chemotherapy with a modified National Protocol for Acute Lymphoblastic Leukemias, adapted from the St Jude Children's Research Hospital TOTXV protocol. However, after the steroid window with prednisone (60 mg/m2), the patient presented hyperleukocytosis, hyperuricemia, elevated lactate dehydrogenase, and alkaline phosphatase. These results were considered poor steroid responses, so the patient was reclassified as high-risk.
First relapse
On November 30 (2013), remission induction was initiated. On November 30 and December 7, the patient received daunorubicin (30 mg/m2); on December 1, 3, 5, 8, 10, 12, 15, 17, and 19, L-asparaginase (10,000 IU/m2), and dexamethasone (6 mg/m2) for 28 days; on December 7, 14, and 24, vincristine (0.05 mg/kg). Two weeks later, an episode of fever was recorded, along with neutropenia and mucositis. The patient was treated with cefepime and amikacin. Bone marrow aspirate (BMA) on day 7 showed 1.5% blasts, and cerebrospinal fluid (CSF) was negative. Blood count on day 8 showed no blasts. BMA on day 21 showed 0.5% blasts and negative CSF. BMA on day 28 showed 0.5% blasts and negative CSF. On January 7 (2014), hematological remission was documented, and the first intensification with etoposide (100 mg/m2 4-hour infusion every 24 hours for 5 days) and cytarabine (1000 mg/m2 1-hour infusion every 12 hours for 3 days) was initiated. Hepatomegaly decreased by 30%.
In February, the first consolidation with methotrexate (2.5 mg/m2) started. During the third week of maintenance, the patient presented a very early relapse to the BM with 37% blasts (Table 1). Immunophenotyping by flow cytometry suggested that the blasts were positive for CD19, CD22, CD79a, CD15, CD33, CD34, and CD45 (Figure 1B). Similar to the diagnosis phase, a normal cytogenetic study (translocations panel) was reported in this relapse. A hematopoietic stem cell transplantation (HPSCT) was proposed to the parents, but they did not accept it. She continued with second-line chemotherapy according to the institutional protocol with five drugs with cytarabine for myeloid markers. BMA on day 14 showed adequate remission. In August (2014), studies for EBV were conducted again, resulting in a past infection. Eight months later, the patient presented with fever and neutropenia; she received a piperacillin/tazobactam regimen for 19 days. Five months later, a CSF was performed, with negative results for infiltration. Treatment continued until week 98 of maintenance without complications.
Second relapse (lineage switch)
On August 14, 2017, the patient presented to the hospital with petechiae. Laboratory data revealed WBC 43,000/μL, 18% blast cells, 6.5 g/dL hemoglobin, and a platelet count of 19,000/μL (Table 1). The BM was hypercellular and had 30% blasts. Immunophenotyping showed positivity for CD45, CD34, CD117, HLA DR, MPO (myeloperoxidase), CD13, and CD33 (Figure 1C). The patient was diagnosed with acute myeloid leukemia at the age of 5 years. At this point, therapeutic options were explained to the parents considering that chemotherapy would no longer have curative purposes since the prognosis changed due to the lineage switch and not having received HPSCT after the first very early relapse. The parents opted for palliative care; unfortunately, the patient died two months later at home. The family did not authorize an autopsy.
Flow cytometry
Flow cytometry was performed on BMA samples. Samples were processed according to the standard protocol. Mononuclear cells were stained with fluorochromes and antigens from Becton Dickinson Biosciences®, San Diego, CA. Samples were acquired on a FACSCalibur cytometer (Becton Dickinson Biosciences) and subsequently analyzed with CellQuest software (Becton Dickinson Biosciences). Positive antigens were defined as those with fluorochrome expression ≥ 30% of the cell population expressed the fluorescence marker above the cut-off point, using the corresponding isotype control. BM had 14.5% blasts. Flow cytometry showed a population of precursor B cells with aberrant myeloid markers expressing CD45+, CD19+, CD22+, CD79a, sKAPPA+, sLAMBDA+, CD10+, and MPO+ (Figures 2A-2C). Conventional cytogenetic analysis revealed a 46 XX karyotype; the translocation panel was negative for t(9;22) (q34;q11), t(5;14) (q31;q32), t(12;21) (p13;q22), t(1;19) (q23;p13.3), and 11q23 (MLL). No variations were detected in NRAS, KRAS, PHF6, JAK-STAS, RAS, NOTCH-1, or PAX5 genes. Leukemia chimeric gene screening was negative for BCR-ABL. The IKAROS transform study was not performed. Fluorescence in-situ hybridization (FISH) was negative for MLL rearrangements.
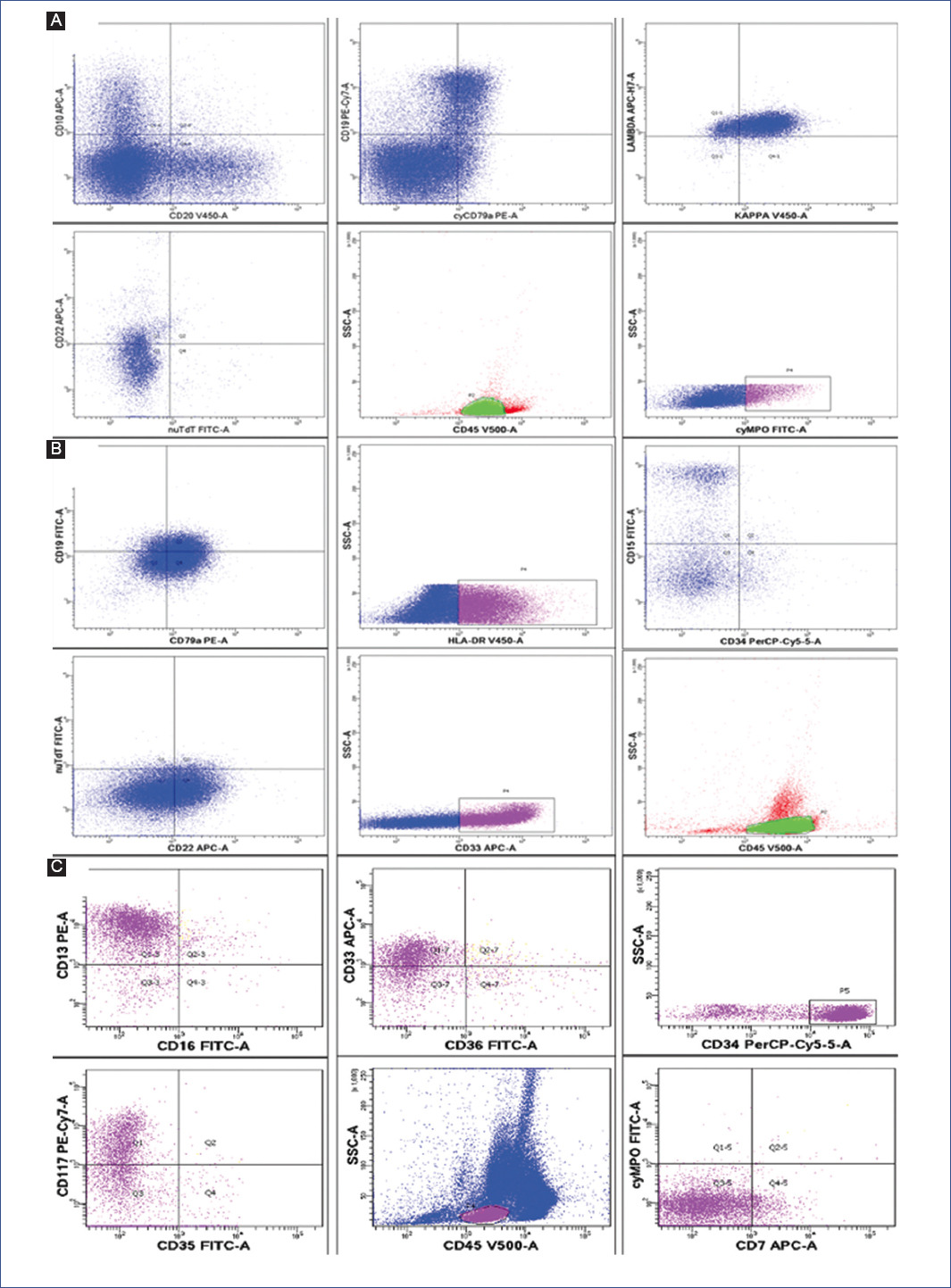
Figure 2 Flow cytometry at different stages of treatment. A: diagnosis (CD45+, CD19+, CD22+, CD79a+, sKAPPA+, sLAMBDA+, CD10+, and MPO+). B: first relapse (CD19+, CD22+, CD79a+, CD15+, CD33+, CD34+ and CD45+). C: second relapse (CD45+, CD34+, CD117+, HLA DR+, MPO+, CD13+ and CD33+). MPO: myeloperoxidase.
Myo1g test
We measured the expression of Myo1g by IF and qRT-PCR at diagnosis, during the poor response to the steroid, and at first relapse. For the second relapse, it was not possible to obtain the sample. As a control, we used a cohort of hematologically healthy children, as recently reported7. Briefly, peripheral blood mononuclear cells (PBMC) were isolated by density gradient with Lymphoprep (Axis 250 Shield). Samples were divided into two parts: one was stored in Trizol for mRNA isolation, and the other was used for IF. Samples were placed on slides, fixed with 4% paraformaldehyde for IF, and processed as indicated7. We quantified the fluorescence intensity of at least 30 cells at diagnosis and different phases of treatment. Images were analyzed using Fiji, Image J software (NIH)18.
qRT-PCR
RNA was extracted from PBMCs using the RNeasy mini kit (QIAGEN) and reverse transcribed into cDNA using the Quantitect Reverse Transcription kit (QIAGEN).
Myo1g expression was measured by quantitative PCR using the Agilent Mx3005 P thermocycler, Universal Probe Library (Roche), with a set of Myo1g-specific primers. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. Light Cycler 480 master mix (Roche) was used for amplification. Amplification was carried out at 95°C/10s, 56°C-59°C/30s, 72°C/11s for 45 cycles. Fold change values of gene expression were calculated with the 2-ΔΔCt method using the mean of triplicate measurements7. Myo1g expression at diagnosis was moderately high in bone marrow (BM) compared to controls (Figure 3A, 3B). However, at the end of the steroid window (Figure 3C) and during the first BM relapse (Figure 3D), significant overexpression was observed by qRT-PCR and immunofluorescence (Figures 3E and 3F), respectively. The sample from the second relapse could no longer be collected because the patient entered palliative care, and the parents no longer allowed further studies.
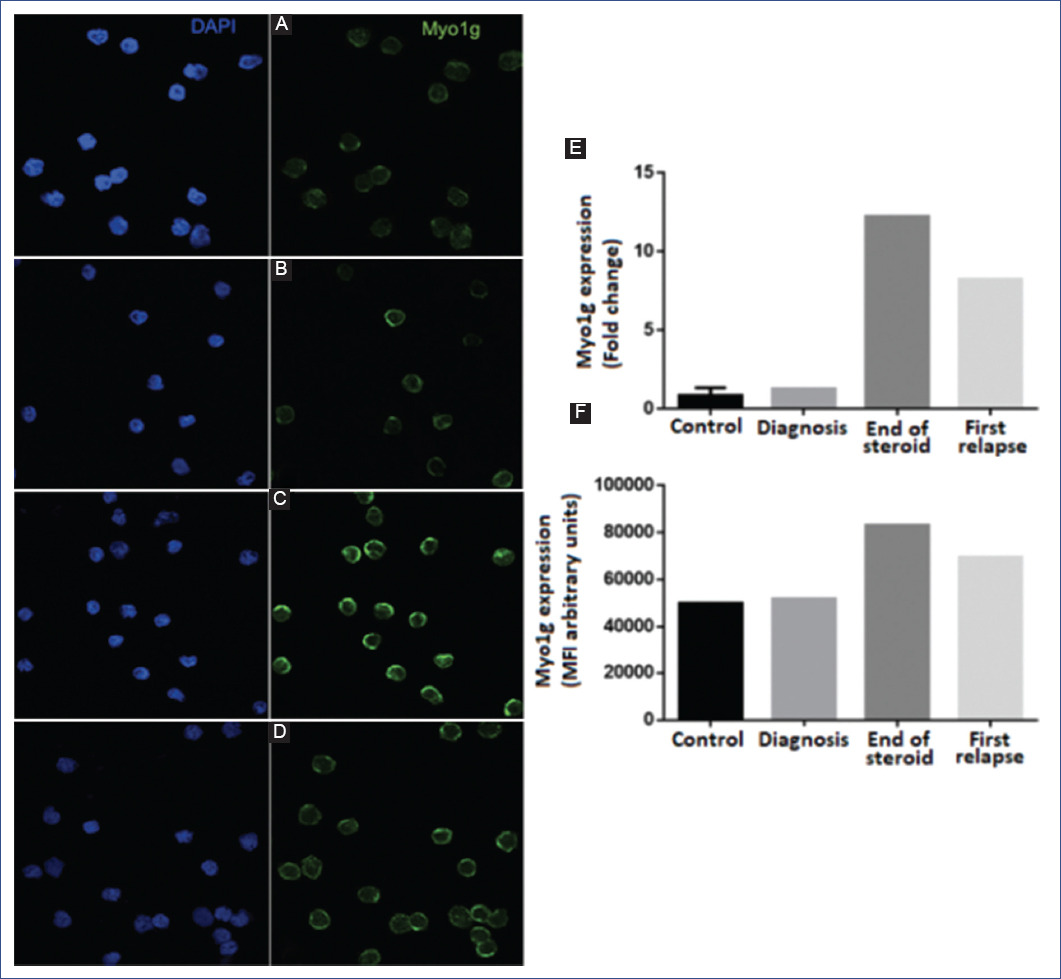
Figure 3 Expression of myo1g at different stages of the disease. Representative confocal images of the expression of Myo1g in A: control; B: diagnosis; C: end of steroid window; D: first relapse; E: expression of Myo1g by qRT-PCR (real-time quantitative reverse transcription polymerase chain reaction) expressed as fold change; F: quantification of Myo1g expression of at least 50 cells, measured by fluorescence intensity (arbitrary units).
Discussion
The incidence of ALL is higher in Latin America than in other parts of the world, with rates of up to 120 patients per million per year19,20. Therefore, it is likely that patients with ALL in this region present biological variations compared to other areas. As a result of epidemiological studies21, it is known that 1-9-year-old female patients have better survival than those outside this age range, such as our patient. At the same time, the prognosis is unfavorable for patients with a central nervous system (CNS) infiltration and leukocytosis (> 50,000/mm3) at diagnosis22. On this basis, in addition to the response to chemotherapy and the leukemia phenotype, the patient's risk of relapse could be established23.
In general, leukemia patients with BM relapse show decreased survival. Moreover, with further complications such as a second BM relapse, myeloid lineage switch, and liver as the primary origin of leukemia, the possibility of cure becomes < 10%. Therefore, third-line chemotherapy could be administered in these cases, but not for curative purposes. Also, the possibility of a third relapse is latent, and secondary toxicity due to chemotherapy could cause organic, infectious, and severe bleeding that would increase mortality. Another possibility is a refractory state of leukemia that would require transplantation. However, this procedure does not have a good prognosis after a second BM relapse. At the time of the second relapse, our patient was not in remission, had tumor activity, and a 100% compatible donor was unavailable. Therefore, the survival prognosis was very poor. Moreover, neither a new chemotherapy cycle for curative purposes nor the HPSCT procedure could be guaranteed, so there were no curative options from the oncological point of view. Therefore, palliative care was the best option to maintain the patient's comfort, seeking the best quality of life.
The steroid window is part of the initial treatment in ALL and is used to assess drug response as a prognostic factor24. In this case, the patient was staged as LR; however, Myo1g overexpression at the end of the steroid window and the increase in leukocytes showed in BC served as biomarkers of HR and potentially as a biomarker of relapse. This report suggests using Myo1g as a biomarker to aid in the classification of patients who meet the characteristics of LR to HR from the onset of the disease; consequently, this will help to start treatment in a targeted and timely manner, thus potentially improving patient survival. Leukemic transformation and clonal expansion of ALL can occur at different stages of the lymphoid maturation and differentiation25,26, making the course of the disease even more complex, as in this case. Not all cases of ALL express antigens of a single lineage27; there are cases of ALL expressing associated myeloid antigens (My + ALL), such as our patient, and cases of acute myeloid leukemias expressing associated lymphoid antigens (Ly + AML)28. In contrast, acute leukemias of mixed lineage (ambiguous lineage) represent a heterogeneous group of rare and poorly differentiated leukemias with features of both lymphoid and myeloid lineages.
The diagnosis of BCP-ALL represents a challenge considering the new classifications, such as the WHO 2017 classification. In this case, the patient initially presented BCP-ALL expressing a myeloid marker (MPO) and negative CD14, so mixed phenotype acute leukemia (MPAL) was considered a differential diagnosis as MPO was the only myeloid marker weakly expressed29.
New subtypes of BCP-ALL with lineage-switching have been described. Novakova et al. found an early switch to monocytic lineage and loss of the B-cell immunophenotype, including CD19 expression30; therefore, more complexity is added to diagnosing this type of neoplasm. The presence of monocytosis (4%) supports the diagnosis. Moreover, a decrease in lymphoid blasts but not in myeloid blasts was observed with chemotherapy, which from the first relapse already showed CD15 + and CD33 +. The expression of myeloid markers was unclear compared to lymphoid blasts, so the diagnosis was agreed as lineage switch and not MPAL. Since this topic may be debated and discussed, it should be supported with cytogenetic studies for better staging and treatment.
Some conditions could explain the lineage switch at relapse; one is the possibility of a second neoplasm after two or three years of treatment with high doses of etoposide. However, this is more frequent when the initial neoplasm is of myeloid phenotype. Another hypothesis to explain the changes in the immunophenotype during relapse is clonal selection15. A previous study showed that MLL rearrangements were prevalent in almost 80% of pediatric patients with B-ALL who underwent lineage conversion to AML after chemotherapy with or without hematopoietic stem cell transplantation11. These rearrangements were negative in the present case.
Currently, there is no standard recommended therapy for lineage-switching leukemia. A case report of an infant with lineage-switching leukemia showed that allogeneic hematopoietic stem cell transplantation (alloHSCT) as consolidation therapy after remission resulted in a well-controlled disease without relapse over 2 years31. In the present case, alloHSCT was not feasible due to the patient's advanced condition and lack of remission since the first diagnosis.
Myo1g overexpression was partially evident at disease onset but increased upon poor steroid response and remained elevated during the first relapse. Therefore, Myo1g is proposed as a relapse biomarker. These data suggest de novo myeloid leukemia after lymphoid leukemia as another differential diagnosis. However, Myo1g was not measured in the second relapse because the parents no longer authorized invasive procedures. Thus, the definitive diagnosis was lineage switch.
In summary, we described a pediatric case of refractory B-ALL with lineage conversion to AML that exhibited alterations in Myosin 1g expression. Further studies are required to investigate the significance of Myo1g expression in leukemia. Finally, targeting multiple antigens on leukemia-initiating cells may be a better strategy to reduce the likelihood of lineage switch.

