











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1 Introduction
Osteoporosis, characterized by low bone mineral density (BMD) that causes susceptibility to fracture, is related to a decrease in the production of dehydroepiandrosterone (DHEA), an adrenal steroid (Cormier et al., 2001; Zhang et al., 2014) and cholesterol derivative (Baeza-Jiménez et al., 2014). In humans, DHEA is synthesized in the adrenal cortex, gonads, brain and gastrointestinal tract, and may be produced by other organs like heart (Nakamura et al., 2004). DHEA affects the endocrine, immune and metabolic systems (Bellino et al., 1995) due at its antioxidant effect as well have it some natural molecules called polyphenols to promote health benefits (Sánchez-Rangel et al., 2013). It has been reported that DHEA has an anti-inflammatory role in endothelial cell veins (Gutiérrez et al., 2007).
In humans, DHEA levels decline with aging (Nestler et al., 1988; Macewen and Kurzman, 1991). Low serum levels of DHEA are associated with an increased risk of age-related chronic diseases like insulin resistance (Schriock et al., 1988), obesity (Nestler et al., 1988), cardiovascular disease (Macewen and Kurzman, 1991), cancer (Barrett- Connor et al., 1986), immunodeficiency (Schwartz et al., 1986), and psychosocial problems such as depression and/or a general deterioration in the sense of well-being (Casson et al., 1993).
Regenerative medicine is currently looking for new strategies to improve the cell therapies for osteoporosis by using mesenchymal stem cells (MSCs) for bone repair. MSCs have been isolated from different tissues, those from human bone marrow (hBM) being the easiest to harvest and expand in vitro. In this context, human bone marrow is the niche of hematopoietic (Andrade-Zaldívar et al., 2014) and mesenchymal stem cells, the latter of which are multipotent progenitor cells that can differentiate into osteoblasts, adipocytes, chondrocytes, tenocytes, skeletal myocytes and neurons (Rodríguez-Pardo et al., 2010; Wang et al., 2012), which is very attractive for cell therapy. There is some evidence that different concentrations of DHEA can induce osteogenic differentiation of hBM-MSCs (Kaivosoja et al., 2010). When administrated in hormonal replacement therapy, DHEA improves BMD (Morales et al., 1998; Baulieu et al., 2000), but how DHEA do it, remains poorly studied. The aim of the present study was to determine the effects of DHEA on osteogenic gene expression in mesenchymal stem cells from the bone marrow of healthy patients. The results support that DHEA is suitable for use as a co-adjuvant in the strategies for alternative treatments in bone repair.
2 Experimental details
2.1 Materials and methods
2.1.1 Human bone marrow mesenchymal stem cell isolation
Three samples of human bone marrow (hBMs) were donated by the orthopedic service of the Hospital General de Puebla SSA. hBMs were diluted 1:2 in Hanks balanced salt solution (HBSS; Gibco). A mononuclear pellet (MNCs) was recovered by density- gradient centrifugation (Lymphoprep 1.077 g/mL) and cultured in 25 cm2 flasks (BD Falcon, Becton Dickinson) at a concentration of 30 × 106 nucleated cells in 5 ml of Dulbecco's modified Eagle's and F12 medium (DMEM), glutamax 1X, 15% fetal bovine serum, and antibiotic-antimycotic 1X (Gibco, USA). Cultures were maintained at 37°C in a 5% CO2 atmosphere and after 72 hours non-adherent cells were removed. When 70%-80% of adherent cells reached confluence, they were trypsinized (0.05% trypsin/EDTA) at 37°C for 5 to 10 minutes, then harvested and expanded in a Petri dish 100 x 20 mm (Corning, USA). A homogeneous cell population was obtained after 2 to 3 weeks of culture. Cell cultures at passage 3 were used for all experiments.
2.1.2 Monoclonal antibodies and immune phenotyping
Monoclonal antibodies against human CD45 conjugated to fluorescein isothiocyanate (FITC), CD34 conjugated to FITC, CD13 conjugated to phycoerythrin (PE), CD105 conjugated to FITC, CD73 conjugated to PE, and CD90 conjugated to FITC were purchased from Biolegend (England). Monoclonal antibodies against human CD14 conjugated to PE as well as HLA-DR conjugated to FITC were purchased from Santa Cruz, CA, USA. Cells were washed twice with ice-cold phosphate- buffered saline (PBS) supplemented with 1% bovine serum albumin (BSA), followed by incubation with saturated concentrations of the appropriate antibodies for 20 minutes at 4°C. Afterwards, cells were washed twice in ice-cold PBS/1% BSA and fixed with 1% paraformaldehyde in PBS. Samples were acquired through FacScalibur (Becton Dickinson) and were analyzed by the Cell Quest software program.
2.1.3 In vitro differentiation assays
hBM-MSCs were plated at 2,000 cells/mL and then the differentiation medium was added. To induce osteogenic differentiation, the cells were cultured for 21 days in the presence of StemPro osteocyte/chondrocyte differentiation basal medium with StemPro osteogenesis supplement or StemPro adipogenesis differentiation basal medium with adipogenesis StemPro supplement (Gibco, USA). Then cells were fixed with formol at 10% and were stained with specific dyes (see the following section).
2.1.4 Evaluation of osteogenic and adipogenic differentiation potential
Osteoblasts were identified by staining with Alizarin Red S (Sigma). Briefly, the hBM-MSC cultured at 21 days with StemPro osteogenesis medium and were washed with PBS and incubated with 2% Alizarin Red S solution (pH of 5.5 with 0.5% NH4OH, and that identifying deposits Ca++ extracellular) for 5 min before being observed in an optical microscope (Leica, Germany) at 10X and 40X objective with integrate digital camera DP24 U/S 3016433.
For confirm that hBM-MSC differentiate into osteoblast was performed semi-quantification assays. After hBM-MSC were Alizarin red staining they were PBS washed and 1 ml phosphate buffer (8 mM Na2HPO4 + 1.5 mM KH2PO4) containing 10% cetylpyoidimium chloride (Sigma) was added and this was incubated for 10 min to dissolve the alizarin red dye. Later this supernatant was measured spectrophotometrically at 550 nm with a microplate reader (BioRad, USA).
Adipocytes were identified by Oil Red staining (Sigma), Briefly, the hBM-MSC were cultured with StemPro adipogenesis medium for 21 days and were staining with Oil Red (for identifying intracellular lipid vesicles, Sigma) for 15 min at room temperature. Afterward, cells were washed completely with isopropanol (Sigma) at 60% and then observed in an optical microscope (Leica, Germany) at 10 and 40X objective with integrate digital camera DP24 U/S 3016433.
2.1.5 Statistic analysis
Values are expressed as mean ± standard deviation. Statistics analyses were performed with students's t test and one way ANOVA by Microsoft Excel. A significant difference was considered at p ≤ 0.05.
2.1.6 Genetic expression and reverse transcription polymerase chain reaction
Total RNA was isolated from the hBM-MSC using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Afterwards, the total RNA sample was used for synthesis of cDNA, according to the manufacturer's instructions (Illumina, USA). Primers were designed by using the Oligo Explorer 1.0 program with the following sequences: forward-GAPDH (5' - TCAACGACCACTTTGTCAAG-3') and reverse- GAPDH (5' -ACTGTGAGGAGGGGAGATTC-3'), forward-SPARC (5'-CAGAGGAAACCGAAGAGG AG-3') and reverse-SPARC (5'-GCAAAGAAGTGG CAGGAAG-3'), forward-RUNX2 (5'-AGTGCGGTG CAAACTTTCTC-3') and reverse-RUNX2 (5' - CTGCTTGCAGCCTTAAATGA-3'), forward-BMP-2 (5'-GGACGCTCTTTCAATGGAC-3') and reverse- BMP-2 (5' -GGTGGGTCTCTGTTTCAGG-3'). Gene sequences were obtained from the database (http://www.ncbi.nlm.nih.gov/pubmed) and specificity was analyzed by BLAST. To carry out RT-PCR endpoint for GAPDH, BMP2, SPARC and RUNX2 genes, an Eppendorf Mastercycler Thermal Cycler® was used for PCR amplification: denaturation at 94 °C for 1 min; alignment at 58-60 °C for 1 min; extension at 72 °C for 30 sec, 30-35 cycles. The reaction products were resolved by electrophoresis on 2% agarose gel dissolved in TAE buffer solution (40 mM Tris-acetate and 1mM EDTA) and visualized with ethidium bromide staining in a documentation system GEL DOC XR (BioRad).
3. Results and discussion
3.1 Cell morphology and immunophenotyping
hBM-MSCs are a morphologically distinct population in primary culture. Although the cell population tends to become visually more homogenous with subsequent in vitro expansion (Nestler et al., 1988; Schriock et al., 1988; Macewen and Kurzman, 1991), the cells must be characterized under the protocol of the International Society for Cell Therapy (ISCT). hBM-MSCs grew as a monolayer of plane and larged cells, and in confluency they assumed a more spindle-shaped homogenous population with a fibroblast-like morphology at pass three (Fig. 1A). To evaluate plasticity, hBM-MSCs were differentiated into osteoblasts and adipocytes for 21 days. Lipid vesicles (droplets) were found in mature and partialmature adipocytes cells-hBM-MSC differentiated (Fig. 1B-C) and extracellular Ca++ deposits were identified in osteoblasts-hBM-MSCs differentiated (Fig. 1D-E).
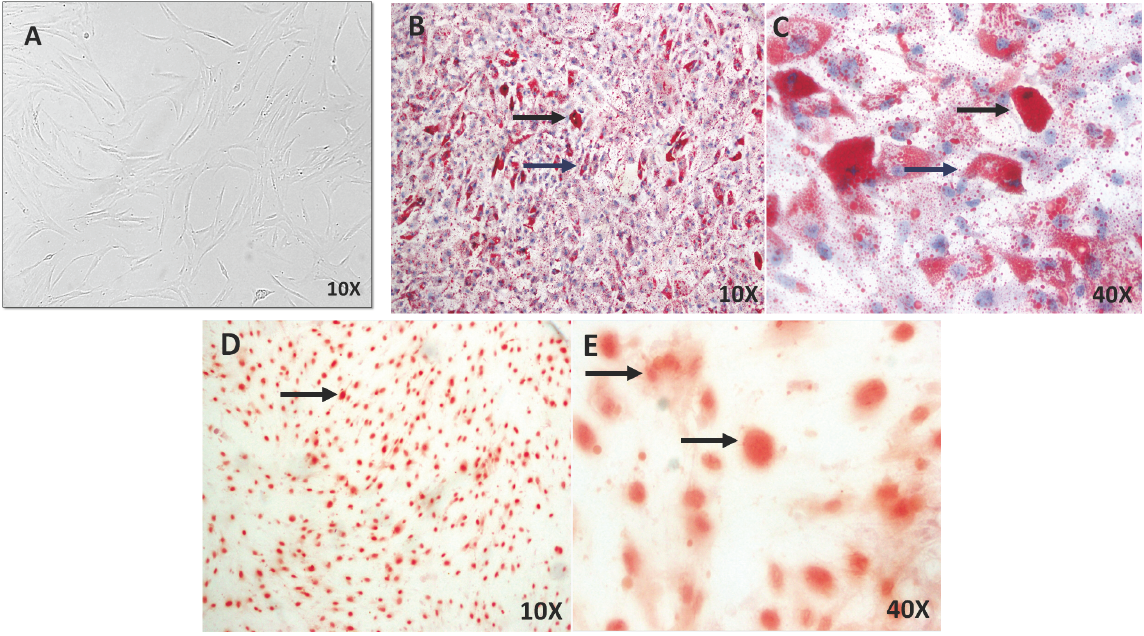
Fig. 1 Cell cultures of hBM-MSCs and characterization of cell plasticity in vitro. A) hBM-MSCs in culture shows large, flat cells, spindle-shaped and fibroblastic morphology at pass three. B-C) Mature (black arrows) and partial- mature adipocytes (blue arrows) were identified by Oil red stainings for lipid vesicles in hBM-MSCs differentiated at 21 days. D-E) Osteoblast were identified by Alizarin red stainings for Ca++ deposits in hBM-MSCs differentiated at 21 days (arrows). Microscopy Ligth 10X and 40X.
Another standard proposed by ISCT is a general panel of positive and negative cell surface markers that now are commonly used to characterize the immunophenotypeofMSCs(Rodríguez-Pardoetal., 2010). A wide panel of cell-surface antigens was measured by flow cytometry analysis. hBM-MSCs had diminished expression of hematopoietic markers: CD14 (1.23%), CD34 (1.47%), CD45 (1.76%) and HLA-DR (2.05%). However, they were positive for CD13 (54.1%), CD73 (47.91%), CD90 (51.75%) and CD105 (15.34%) (Fig. 2). These results indicate that population of hBM-MSCs was heterogeneous and poorly enriched, but they accomplished the ISCT standards in accordance to reports in literature. (Phinney et al., 1999; Gnecchi and Melo, 2009).
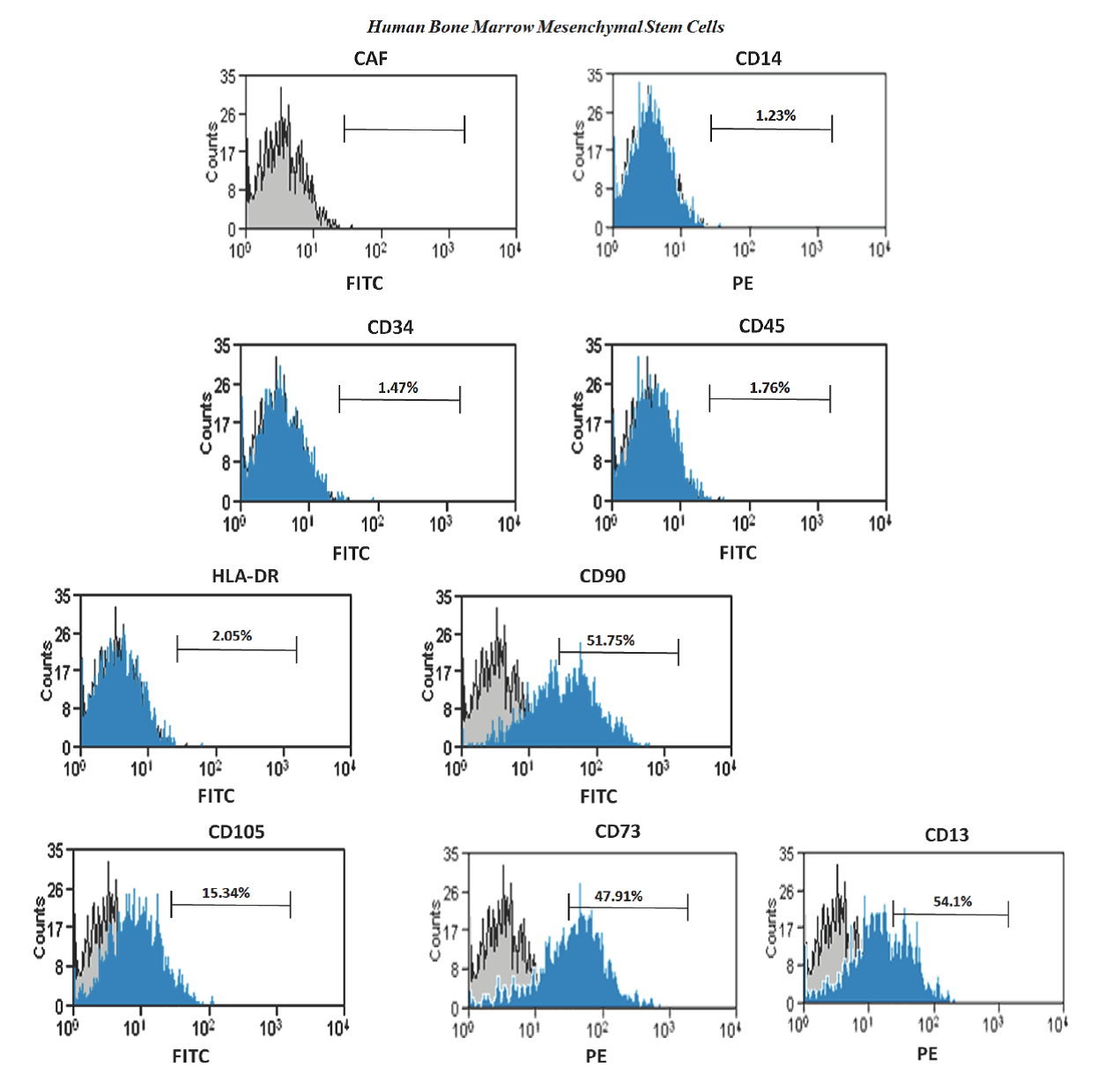
Figure 2 Flow cytometric analysis of membrane markers on hBM-MSCs at passage three. The hBM-MSC demonstrate that were negative for hematopoietic cell markers CD14, CD34, CD45 and HLA-DR and CD13, CD73, CD90 and CD105 were still expressed. Antibogy anti-IgG1 were used as control (CAF). FITC, fluorescein isothiocyanate; PE, phycoerythrin.
hBM-MSC used in this study showed low percentages of membrane markers, but this may be due to many factors, for example, Flores et al. (2006), mentioned that depending on the methodology used this may influence to obtain a population of hBM-MSC either pure or homogeneous. Using ficoll-hypaque cell population results less pure and more heterogeneous, and when other sophisticated methodology is used or a combination of them, like works by Neagu et al., (2005) and Huang et al. (2015), who used separation method by ficoll-hypaque and then recovering cells were sorted using fluorochrome labeled antibodies, the population isolated resulting was pure and homogeneous. However, that results were on passage 25, when the population is fairly homogeneous and 99% CD105 positive. The above mentioned is consistent with observations by Mafi et al., (2011), where variability in expression of membrane markers may be due to the state of cell proliferation and culture at the time to be evaluated. Results shown here were carried out in population of hBM-MSC at passage three and only were purified using ficoll-hypaque gradient. It would be interesting that cells used in this work could be evaluated in higher passages and for other membrane markers presents also in adherent cells from bone marrow, such as endothelial and/or epithelial cells.
3.2 Gene expression in hBM-MSC stimulated with DHEA
GAPDH (housekeeping) remained constant under all conditions, while the RUNX2 and SPARC mRNA expression had basal levels in the control cells and higher expression with DHEA treatment at each of concentrations. (Fig. 3). Hence, it is possible that the role of RUNX2 and SPARK genes are not directly related to osteogenic differentiation induced by DHEA. Niu et al., (2012) described the basal mRNA expression of RUNX2 in WRO and TPC-1 cells and showed that this is influenced by fetal bovine serum (FBS) similar to results here presented.
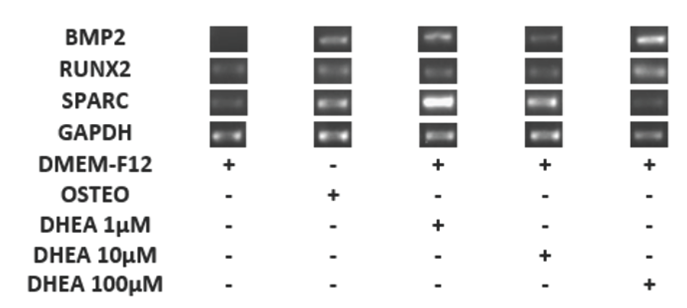
Fig. 3 Differentiation of hBM-MSC to osteoblastic cells for with DHEA at 7 days. The hBM-MSC were cultured in absence (-) or the presence (+) DHEA at 1, 10 and 100 (M or osteogenic medium (osteo) during 7 days and afterwards RT-PCR was carried out. The expression of GAPDH (endogen control), RUNX2, SPARC and BMP2 were observed in a 2% agarose gel. The DHEA induced the expression of BMP2 at 1, 10 and 100 μM on hBM-MSC similar to osteogenic medium.
It is known that both gene expression and protein synthesis of osteonectine (SPARC) and RUNX2 are associated in a closely and dependent way (Ducy et. al., Jang et al., 2012). Also it has been reported recently in hBM-MSC a basal expression of SPARC (Karaoz et al., 2009), which is in consistent with the observed in hBM- MSC with or without DHEA treatment (Fig. 3). In contrast with the observed for SPARC or RUNX2, BMP2 mRNA was not expressed in cells without DHEA stimuli (negative control), but it was presented in cells cultivated with osteogenic medium (positive control) as well in DHEA at 1, 10 and 100 μM (Fig. 3). The gene BMP2 is considered one of the most important in relation to the osteogenic differentiation of MSCs (Davis et al., 2011; Quing et al., 2012; Wang et al., 1990; Wozney et al., 1988) because it′s expression orchestrate other osteogenic genes like RUNX2 and SPARC (Jang et al., 2012). In this work RUNX2 expression was observed in all hBM- MSC, possibly because extra to be a gene involved in the osteogenesis process, it could participate in other functions, for example on cell cycle (Ko et al., 2011; Komori, 2010; Chung et al., 2005). Here, results showed in cultures treated with DHEA that BMP2 gene expression was constant (Fig. 3) and similar to observed with osteogenic medium treatment.
3.3 Extracellular calcium resulting from DHEA-induced osteoblastic differentiation
To confirm that DHEA promotes differentiation from hBM-MSC to osteoblasts, hBM-MSC were cultured in presence of osteogenic medium (positive control) or DHEA (1, 10 and 100 μM). The concentration of extracellular Ca++ was much higher in hBM- MSCs exposed to DHEA at 1 μM than hBM-MSCs in other culture media (osteogenic medium or medium with DHEA at 10 or 100 μM; Fig. 4). These results are similar to founded by Kaivosoja et al., (2012), where DHEA at 100 μM induces the synthesis of alkaline phosphatase, osteopondin, osteocalcin and RUNX2. Malik et al., (2010) investigated in hBM- MSC stimulated with 0.1, 1 and 10 μM of β-AET, one metabolite of DHEA, that after 15 days of stimuli the osteoblastic differentiation occurred, which it was corroborated by osteopondine positive cells.
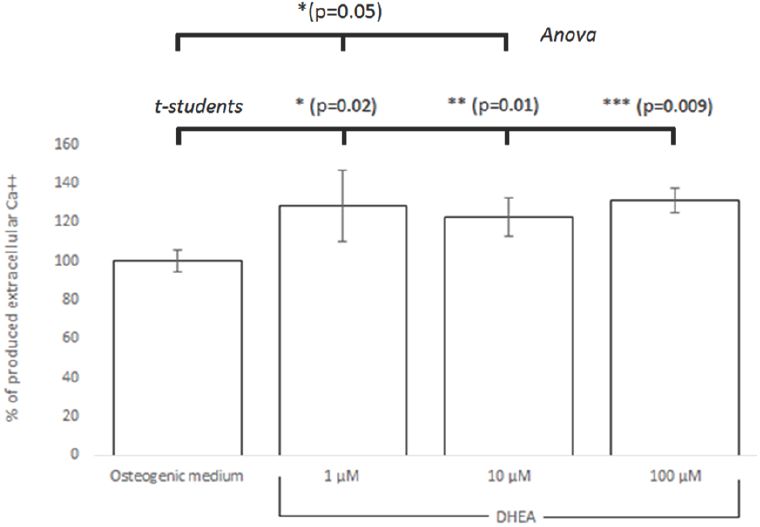
Fig. 4 Semi-quantification of extracellular Ca++ assessed by Alizarin red solubilized in hBM-MSC differentated on osteoblast with DHEA. hBM-MSC were stimulated seven days with osteogenic médium or DHEA at 1, 10 and 100 μM and it is confirmed that DHEA at 1 μM induced the differentiation to osteoblasts. Significant differences were from osteogenic medium (control) are indicated as follows: * (p=0.02), ** (p=0.01), *** (p=0.009) by the Student's t-test and * (p =0.05) by Anova.
Results showed statistical significance between hBM-MSCs cultured with osteogenic medium and 1, 10 and 100 μM of DHEA stimuli analized by students's t test (p = 0.02, 0.01, 0.009 respectively) while data analysis by ANOVA indicates just a significant difference between hBM-MSC osteogenic medium and 1 and 10 μM of DHEA stimuli (p = 0.05).
Concentrations on serum DHEA varies throughout of life reaching a peak at around 10 μM in blood (Jean-Pierre et al., 2013). In this study, physiologic concentrations of DHEA (1 and 10 μM) cells showed an osteogenic differentiation, although the effect is well observed at pharmacological concentrations (100 μM) in comparison to untreated cells. Clinical studies in old subjects (where DHEA keep diminished), physiological DHEA concentrations on blood could be recovered with doses of 50-100 mg of DHEA per day, and the treatment does not present collateral effects (Legrain and Girard, 2003).
These results confirm that DHEA is a good inductor of osteogenic differentiation, and that it could possibly be used alone or as a co-adjuvant in long-term alternative treatments for bone repair. There are some mechanisms which propose how DHEA acts. The Figure 5 shows the proposed signaling for expression of BMP2 induced by DHEA (confirmed in this work), together with information founded in literature and sustained by others authors. DHEA can bind to G-coupled protein receptor [1] (Siddappa et al., 2008) and through of a complex process relationship to G- protein it's activated MAPK signaling pathway [2] (Gavi et al., 2006; Wang et al., 2007). Downstream, some protein kinases like ERK1/2 [3] (Wang et al., 2007) and PKA [4] (Siddappa et al., 2007), could phosphorylate transcription factors as ELK1 [5] (Li et al., 2013) and CREB [6] (Siddappa et al., 2008). In response of this, the expression of BMP2 gene increases [7] (Ionescu et al., 2004; Siddappa et al., 2008), and as a consequence BMP2 is synthetized in cytoplasm. Later on the cell outside, extracellular mineralization [8] (Ronchetti et al., 2013) and bone formation occur [9] (Baulieu et al., 2000; Arlt et al., 2001; Kahn et al., 2002). (Fig. 5).
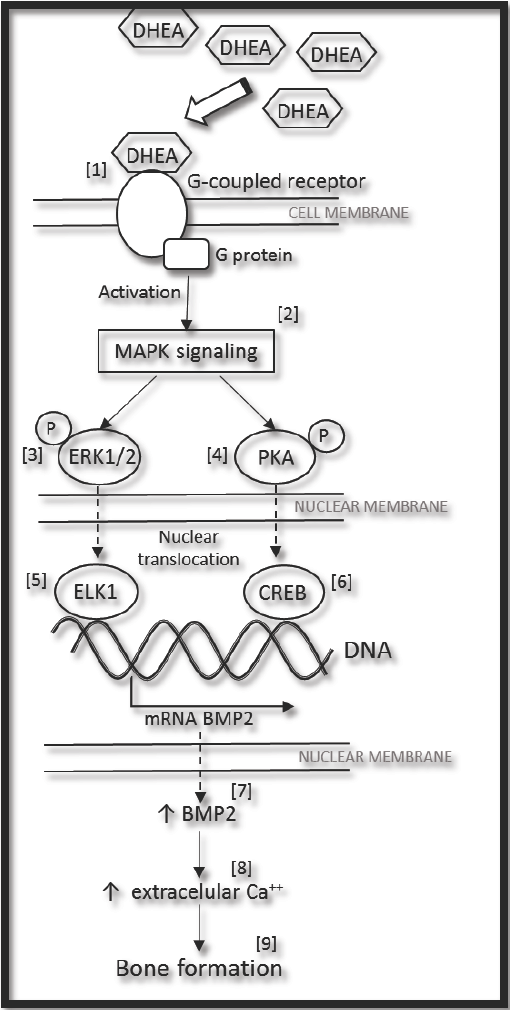
Fig. 5 General proposed mechanism for expression of BMP2 induced by DHEA, (see the text). MAPK (Mitogen-Activated- Protein-kinase); ERK1/2 (Extracellularly-Responsive-Kinase 1/2); PKA (Protein Kinase A); ELK1 (Ets-like protein-1); CREB (cyclic AMP response element blinding protein); and BMP2 (Bone Morphogenetic Protein 2).
Conclusions
The results of the present cultures with hBM- MSCs are indicative of molecular mechanisms of osteogenic dierentiation, according to the standards of International Society for Cellular Therapy (ISCT). DHEA induced osteogenic dierentiation and the principal gene responsible for this result is BMP2. DHEA at 1 and 10 μM physiological doses was found herein to induce high concentrations of extracellular Ca++ as a result of the overexpression of RUNX2, SPARC and BMP2 osteogenic genes, probably by a signaling that involved the molecules and transcription factors from binding DHEA to known receptor. Further studies are necessary to identify specific mechanisms that are involved when using DHEA as a dietary supplement for patients with osteoporosis. At the moment, for these patients, there has been a documented improvement in BMD by induction of osteogenic genes with DHEA treatment.

