











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Amino acids (AA) are the basic units of proteins; if there is any AA deficiency, the synthesis of proteins is altered, causing a unbalance in the cellular physiology and organism. AA have different functions and are essential for the physiology of the body; for example, hormones such as insulin, growth hormone, and glucagon, are made up of AAs. It is known that some AAs regulate the in vitro and in vivo secretion of insulin in pancreatic β-cells. AAs play an important role in different processes related to gene expression, including the modulation of proteins that regulate mRNA1.
In the central nervous system (CNS), some AA are neurotransmitters and have a key role in neuronal communication, such as glutamate and glycine. In the case of GABA, despite that chemical structure, it is not considered like AA, because it does not conform proteins. These neurotransmitters are stored in vesicles inside the presynaptic neuron and released into the synaptic cleft by a Ca2+-dependent mechanism and in response to a depolarizing stimulus. These neurotransmitters generate excitatory or inhibitory responses through binding to their specific receptors in the post-synaptic neuron or even the same presynaptic cell. A malfunction of these neurotransmitter communication systems is associated with different neurological or psychiatric disorders. For this reason, several pharmacological therapies aim to regulate release mechanisms, receptor binding, or neurotransmitter reuptake.
In this review, we will focus on the inhibitory neurotransmitter GABA (g-aminobutyric acid), specifically in the participation of their receptors in different neurodegenerative disorders and the different pharmacological strategies aimed at their regulation for the treatment of these pathologies.
GABA
GABA is the main inhibitory neurotransmitter of the CNS and one of the most abundant neurotransmitters in mammals. It was first described in the early 1900s, while its presence and participation as a neurotransmitter in the mammalian CNS was determined until the 1950s. During the following two decades, many studies established its mechanism of action, as well as its inhibitory activity in the cerebral cortex2. We, currently, know that GABA is distributed in most areas of the brain and participates in 40% of the inhibitory synapses of adult vertebrates.
GABA metabolism
GABA is produced in the CNS, through the decarboxylation of glutamic acid, and catalyzed by glutamic acid decarboxylase (GAD). In general, the enzymatic activity of GAD is regulated by its expression levels and the degree of association with the cofactor pyridoxal phosphate (PLP)3. GAD has two isoforms, GAD65 and GAD67, both encoded by different genes, and different expression patterns, but with similar mechanisms regulating their function4,5. GAD65 is located mainly in the synaptic terminals, where it induces vesicular GABA release in a Ca2+-dependent process. GAD65 dissociates from PLP under physiological conditions and increases its activity depending on the demands of the synaptic release of GABA6. Therefore, the main regulatory mechanism for this isoform is the association with its cofactor and not it is level of expression3.
GAD67 is expressed in the cytoplasm, it is associated with PLP under physiological conditions, and is regulated by its levels of expression. GAD67 is involved in cell metabolic activity and is responsible for most of the GABA synthesis in the brain3. However, it has been suggested that cytoplasmic GABA can be released by a Ca2+-independent mechanism. If this is correct, then GAD67 may participate in inhibitory neurotransmission by activating extra-synaptic GABA receptors.
After GABA release into the synaptic cleft, it is reuptake by the GAT-1 and GAT-3 transporters, these systems are expressed in both neurons and glial cells. GABA catabolism is carried out through the GABA transaminase (GABA-T). GABA-T is dependent on PLP, which, in turn, requires GABA and α-ketoglutarate as substrates, generating glutamic acid, and succinic semialdehyde (SSA) as by-products. SSA can be metabolized into γ-hydroxybutyric acid, which regulates GABAB receptors, or can be dehydrogenated to succinate. When GABA is dehydrogenated to succinate via SSA dehydrogenase, it is incorporated into the Krebs cycle, where it participates in cellular energy metabolism. Many clinical and preclinical researches indicate the presence of GABAergic neurons in regions such as the amygdala, hippocampus, hypothalamus, prefrontal cortex, olfactory bulb, retina, and spinal cord.
GABA receptors
GABA exerts its inhibitory effect through two types of specific receptors called GABAA and GABAB, which show different pharmacological, structural, and molecular differences (Fig. 1). GABAA receptors, or ionotropic, share its structural and functional properties with the ligand-gated ion channels or Cys-loop family, which include glycine (Gly), acetylcholine (ACh), and serotonin (5-HT3) receptors7. The complexity of GABAA receptors lies in the number of subunits they contain, and in the different combinations in which they are assembled. There are also variants generated by splicing or RNA editing8. To date, six subunits have been characterized in humans: six type a, three type b, three type g, and one each of type d, e, p, and q, giving this receptor a high degree of heterogeneity. On the other hand, three subunits r have been characterized, which unlike the others form functional homopentamers with different pharmacological properties. These subunits are considered a subfamily called GABAC. In adult mammals, the most abundant isoform is composed of the subunits α1β2γ2. However, the number of GABAA receptor isoforms expressed in mammals is still unknown9 (Fig. 2).
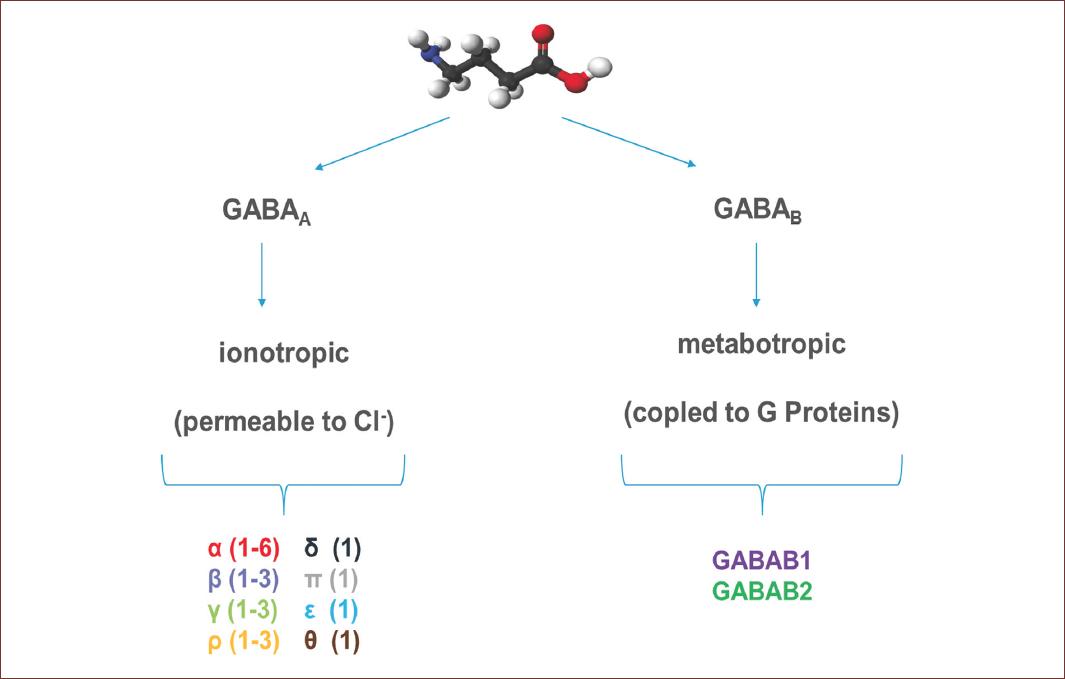
Figure 1 Representative molecular structure of GABA and GABA receptors. GABA is a full agonist of two types of receptors, named according to their mechanism of action. GABAA or ionotropic receptors, are protein channels activated by GABA and permeable to Cl-. These receptors are conformed by different subunits, six type α, three type β, three type γ, and one each of type δ, ε, π and θ. GABAB, or metabotropic receptors, are receptors coupled to G proteins that modulate K+ and Ca2+ channels and are conform for only two subunits. The GABA structure was from https://es.wikipedia.org/wiki/Ácido_γ-aminobutírico (Last accessed on September 20, 2020).
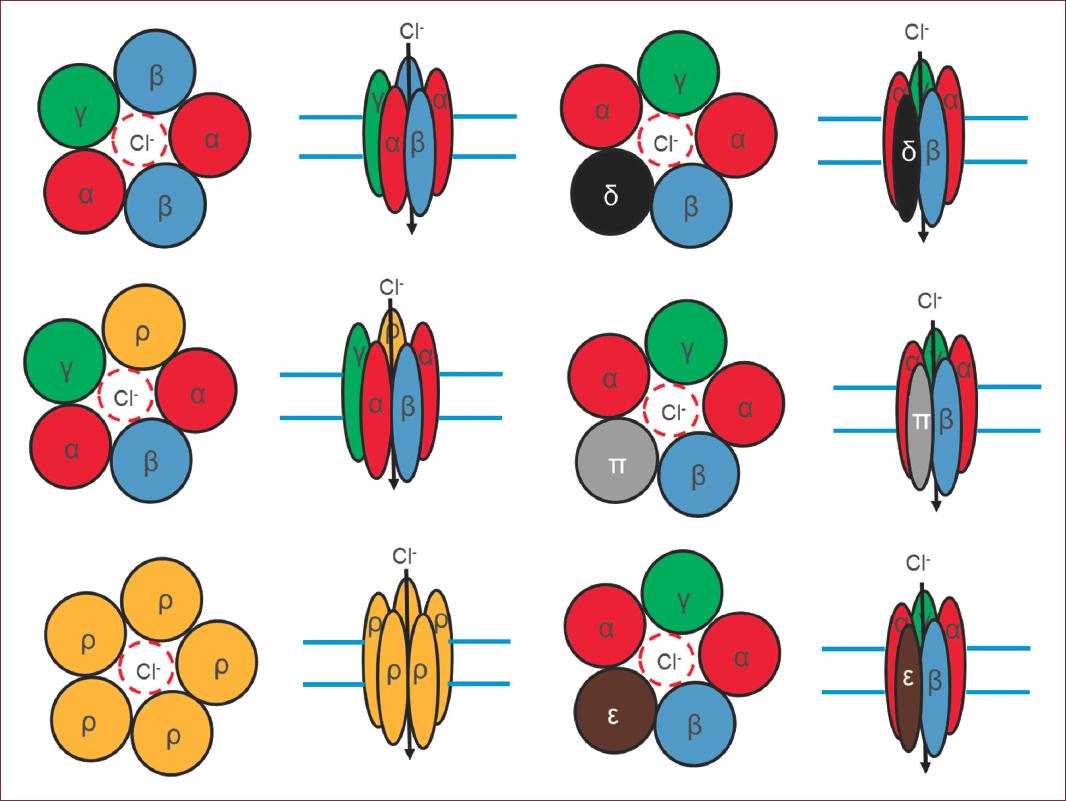
Figure 2 Structural conformation of GABAA receptors. Note the number of subunits that contain, and in the different combinations in which they are assembled. Some of them are variants from splicing. giving this receptor a high degree of heterogeneity. On the other hand, three subunits ρ have been characterized, which unlike the others form functional homopentamers. In adult mammals, the most abundant receptor conformation, composed of the subunits α1β2γ2. However, the number of GABAA receptor isoforms expressed in mammals is still unknown.
GABAA receptors are selectively blocked by bicuculline and picrotoxin, and they are allosterically modulated by neurosteroids, barbiturates, and benzodiazepines. At present, there is a continuously increasing large number of regulatory molecules for this type of receptors, due to their pathological importance10. In the case of GABAC receptors, they are not regulated by modulators and blockers of the GABAA receptor. However, they are sensitive to picrotoxin and 1,2,5,6-Tetrahydropyridin-4-yl)methyl phosphonic acid or TPMPA, and specific GABAC antagonist11 (Fig. 3). GABAA receptors activation leads to the inhibition of synaptic transmission, due to hyperpolarization in response to a Cl− influx through these receptors12. Interestingly, during development there is a delay in the expression of Cl− mobilization systems, generating a high intracellular concentration of this anion13. Therefore, an increase of Cl− conductance in response to the activation of GABAA receptors generates a Cl− outflux current and consequently, cell depolarization, so GABA acts as an excitatory AA during CNS development14.
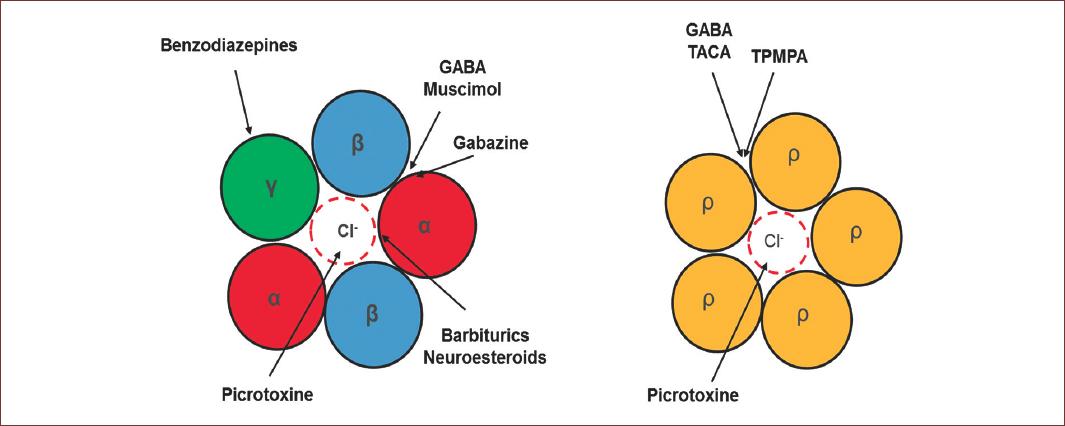
Figure 3 Pharmacology properties of GABAA receptors. At the right is the classical conformation of the GABAA receptor, observe the modulation sites were several drugs neuromodulator act to modulate the GABA physiology. These drugs exert their action in different sites of the receptor. Benzodiazepines in the γ subunit; Picrotoxin actin the pore of the channel and blocked; the Barbiturics and neurosteroids in the α subunit and, Muscimol and GABAzine compete for the GABA binding site located between α and β subunit. On the left side, are functional homopentameric receptor formed by ρ. In this case, Picrotixine, as the α1β2γ2 conformation, acts in the pore of the receptors, however, TACA and TPMA compete for the GABA binding site.
GABAB receptors, or metabotropic, belong to the family of G-protein-coupled receptors or GPCRs, which activate slow responses through the second messengers15, and have a limited structural diversity, unlike GABAA receptors or metabotropic glutamate receptors (mGluRs). GABAB receptors are heterodimers composed of the GABAB1a or GABAB1b subunits combined with the GABAB2 subunit. Nevertheless, despite their poor structural diversity, native GABAB receptors show a varied kinetic and pharmacological response (Fig. 4).
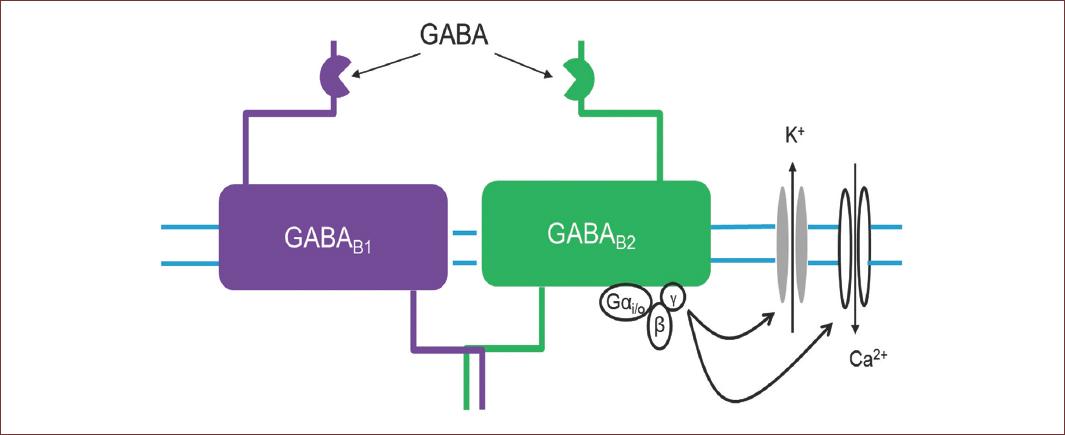
Figure 4 Structural conformation of GABAB receptor. This receptor is heterodimers composed of the GABAB1 or GABAB1, belong to the G-protein-coupled receptors, which activate slow responses through the second messengers, that regulate K+ and Ca2+ channels. Nevertheless, despite their poor structural diversity, native GABAB receptors show a varied kinetic and pharmacological response.
The location of GABAB receptors in the synaptic region is key to regulating neurotransmission. Depending on whether the receptor is presynaptic or postsynaptic, their activation generates an inhibition or disinhibition of synaptic activity. At the post-synaptic level, receptor activation induces a K+ conductance increase, which is responsible for the slow inhibitory events of GABA in the CNS16. The activation of this K+ conductance, coupled with negative regulation of Ca2+ influx at the presynaptic level, decreases the release of GABA by regulating the inhibitory effect mediated by this neurotransmitter17.
GABA and the CNS
The proper functioning of the CNS depends on the balance between the excitatory and inhibitory neurotransmitter systems. The excitatory system is regulated by glutamate, while the inhibitor system is regulated by GABA through interneurons, which modulate the excitatory level generated by glutamate release. These interneurons control the flow of information and the synchronization of the cerebral cortex, despite the existence of a 1:5 proportion with glutamatergic neurons. Suggesting, that the numerical balance between neurons and interneurons determines brain functionality, in the case of the cerebral cortex, the proportion 1:5 indicates that this region is mainly excitatory.
Studies in animal models indicate the presence of GABAergic neurons in regions such as the amygdala, hippocampus, hypothalamus, prefrontal cortex, olfactory bulb, retina, and spinal cord, this observation was corroborated also in human studies. This wide expression of the GABAergic cells indicates that this inhibitory neurotransmitter is involved in many functions in the CNS (Fig. 5), for instance, the thalamocortical pathway, which regulates primordial functions such as behavior, motor control, mood, sleep, and among others.
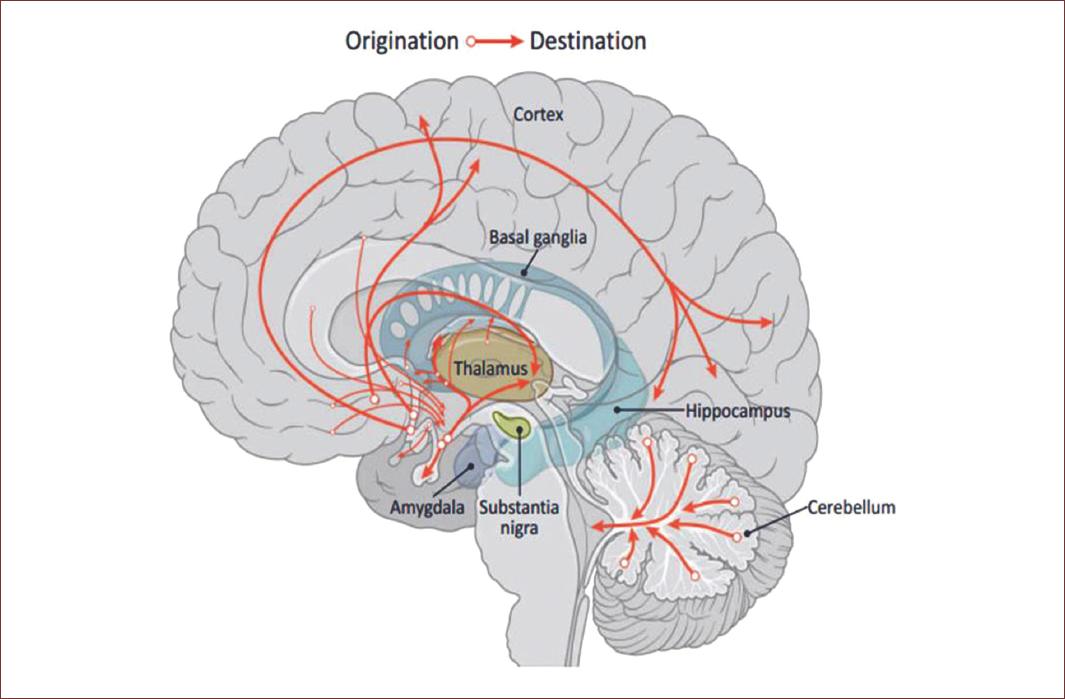
Figure 5 GABA pathways in the brain. The GABAergic system goes throughout the amygdala, hippocampus, hypothalamus, prefrontal cortex, olfactory bulb, including the spinal cord and even the retina. This wide expression of the GABAergic cells indicates the key role of this inhibitory neurotransmitter in functions in the CNS, such as behavior, motor control, mood, sleep, among others. Modified from Brain Chat. https:// thebrainchat.com/photos/gamma-aminobutyric-acid-gaba-is-a-naturally-occurring-amino-acid-that-works-as-a/1243388282530289/.
The thalamocortical pathway consists of GABAergic neurons that project from the thalamic reticular nucleus (nRT) toward the ventral basalis (VB), generating an inhibitory loop for a self-modulation of the nRT. GABAergic neurons of the nRT receive the glutamatergic stimulation from the VB and the cortico-thalamic fibers that are projected from the layer six of the cerebral cortex. In this loop context, GABAA receptors are in glutamatergic thalamocortical and cortico-thalamic neurons, as well as in the GABAergic neurons themselves18.
Thanks to benzodiazepines it has been understood the functional differences between the GABAA receptors located in the nRT and the thalamic relay neurons. Since benzodiazepines increase the neurotransmission regulated by GABAA receptors, these compounds exacerbate absence seizures; however, they show some therapeutic efficacy in clinical and animal models. This double function of benzodiazepines is because they increase the inhibition regulated by GABAA receptors in the nRT, causing a decrease in the inhibitory stimulus of the nRT toward the thalamic relay neurons19. Experimental evidence indicates the participation of GABAB receptors in the pathogenesis of absence seizures20. GABAB receptors regulate K+, activating low-threshold Ca2+ potentials and consequently, burst firing, and the oscillatory behavior of thalamic neurons. Agonists of these receptors exacerbate absence seizures in animal models in a greater proportion than GABAA receptors.
Disorders associated with GABA receptors
Sleep disorders
GABA has important involvement in sleep cycle regulation, and for decades benzodiazepines and gaboxadol (alternative to benzodiazepines) have been prescribed for the treatment of insomnia with certain tolerance; however, these compounds have side effects such as addiction, hallucinations, and disorientation. Several alterations in the thalamocortical pathway are associated with sleep problems, a common symptom in different neurological pathologies, and where it has been described an increase in GABA levels and an altered function of GABAergic receptors that contain the d subunit (d-GABAA)21.
Epilepsy
It is known that the mechanisms regulated by the GABAA receptor participate in the partial or generalized generation of tonic-clonic seizures. Pharmacological and gene expression studies suggest a role in the prevention of seizures by blocking the regulation of GABAA receptors with specific antagonists. Studies point to changes or differences in the expression of subunits of GABAA receptors that correlate with epileptogenesis22. There is a relationship between mutations in the g2 subunit and epilepsy in humans23, similarly, mutations in the gene encoding the d subunit have been associated with some forms of congenital epilepsy in humans24.
The regulation system through the GABAB receptor participates in the prevention of tonic seizures. In humans, an upregulation of the mRNA of the GABAB R1 subunit in the CA1 region, dentate gyrus, and hilus region of the hippocampus has been determined in samples of patients with temporal lobe epilepsy25. A report indicates an increase in antagonist sites per neuron in the CA3 and the hilus regions of the hippocampus in patients with temporal lobe epilepsy26. On the other hand, it was determined that a GABAB R1 polymorphism (G1465A) confers a high susceptibility to temporal lobe epilepsy and increases the severity of the condition27. GABAB R1 knockout mice show several seizures per day, as well as depression-like behavior28.
Anxiety disorders
GABA receptors are of great interest for the treatment of anxiety disorders, due to the therapeutic use of benzodiazepines for these conditions. Tests with positron emission tomography and single-photon emission tomography in patients with panic attacks showed an alteration in benzodiazepine binding sites in different areas of the brain29. Family studies indicate an association between inhibition of behavior and anxiety disorders. Smoller et al. found an association between an intronic polymorphism in the GAD65 gene with anxiety disorders30.
Schizophrenia
There are numerous hypotheses about the pathophysiology of schizophrenia, one of which proposes an alteration of GABA-regulated neurotransmission31. Changes in the expression of genes related to the different subunits that form GABA receptors have been observed32. However, this is not specific for GABA, since there are also changes in gene expression of other neurotransmitters. Another evidence that supports the GABA hypothesis in schizophrenia is that the treatment of this pathology with antiepileptic drugs shows positive clinical results in schizophrenic patients33,34. The use of benzodiazepines in combination with antiepileptic drugs reduces considerably the symptoms of schizophrenia and anxiety. On the other hand, the administration of valproate with neuroleptics controls efficiently irritability symptoms and violent behaviors.
The genes that encode GABAB R1 are located within the loci of schizophrenia, suggesting the participation of this receptor in this disorder35. These observations suggest that the GABAB R1 receptor may be a pharmacological target for schizophrenia.
Depression
In recent years, different reports showed evidence about the association between GABA and depression. For example, a decrease of this neurotransmitter in the cerebrospinal fluid of patients with depression has been reported36. Magnetic resonance spectroscopy in depressive patients showed a reduction in GABA levels, mainly in the occipital cortex (OCC) and in some areas of the prefrontal cortex (PFC)37. Studies indicate changes in the regulation of the genes which encoding GABA receptors. For example, Choudary and col. showed the deregulation of the b3, g2, and d subunits in the frontal cortex38. In this regard, the postmortem analysis by PCR of different brain regions from suicide victims with depression showed alterations in the a5, g1, and g2 subunits of the dorsolateral and lateral inferior PFC39. Fatemi and col. found an increase in the a2, a2, g3, and e subunits in the cerebellum of non-suicidal patients with depression40.
Premenstrual dysphoric disorder (PMDD)
Changes in cognition, mood, and sensitivity to medications throughout the premenstrual cycle have been attributed to hormonal regulation of GABAergic transmission41. PMDD occurs during the luteal phase of the menstrual cycle and is characterized by significant alterations in behavior, mood, and a physical limitation that compromises personal, social, and professional development. In healthy women, plasmatic levels of GABA increase from the follicular to the luteal phase, while they decrease in women suffering from PMDD42. However, in the CNS, GABA levels increase in this same phases43. An increase in cortical inhibitory activity has been reported in healthy women, whereas this effect is not observed in women with PMDD44. Deregulation of GABA levels possibly is secondary to the action of allopregnanolone, a metabolite of progesterone secreted during the luteal phase. This hormone acts on GABAA receptors that contain the a4 and d subunits45. During periods of fluctuation of progesterone concentrations, there are changes in the expression of the a4 and d subunits in the CA1 region of the hippocampus. Furthermore, a decrease in the a1 subunit has been observed42.
Alcoholism
It is well known that the main ethanol target in the CNS is the GABAA receptors46, for example, studies in rodents show that the systemic administration of GABA receptors agonists or antagonists modulates the ethanol consumption47. GABAA receptors increase their probability of aperture and/or their affinity for the agonist after an acute ethanol intake. They are generally potentiated by low concentrations of ethanol (5-50 mM) that correspond to very low intake in humans (a couple of drinks). Chronic exposure to ethanol generates different specific neuroadaptations in GABAergic synapses in different regions of the brain. Furthermore, chronic exposure to ethanol alters the expression of different subunits of GABAA receptors at the transcriptional level in different regions of the brain involved in the development of alcohol dependence. For example, in the cerebral cortex, chronic exposure to ethanol decreases mRNA and the expression of α1, α2, and α3 subunits, and increases the expression of α4, β1, β2, β3, γ1, and γ2 subunits48. In humans, sequencing assays have identified different genes encoding GABAA receptors associated with alcohol consumption, such as α3 (GABRA3) in the prefrontal cortex, γ1 (GABRG1), and γ2 (GABRG2) in the hippocampus49. These expression changes probably alter the assembly of GABAA receptors, modifying their binding affinity to the ligand and, consequently, their function. In the case of the hippocampus, alterations in the expression of GABAA receptors depend on the time of exposure to ethanol. A 40-day chronic exposure significantly increased the levels of the α4 subunit; however, no alterations were observed in the α1, α2, α3, β (2/3), or γ2 subunits50. These results indicate a regulation in the expression of subunits of GABAA receptors specific to each region of the brain that is dependent on the length of exposure to ethanol.
In the case of GABAB receptors, they are determinants of the specificity of ethanol effects on certain regions of the brain. For example, Peris and col. showed that chronic ethanol treatment increases the release of GABA, decreasing long-term potentiation51. Exposure to baclofen (GABAB antagonist) decreases this release of GABA, whereas 2-OH-saclofen (GABAB antagonist) enhances it. This chronic effect of ethanol on the release of GABA into the synaptic cleft could explain the reduction in long-term potentiation observed after chronic exposure to ethanol.
Conclusion
Its complex structural diversity, associated with its differential expression pattern in the neural loops of the brain, makes it very difficult to understand its inhibitory control in different neural networks. Although our knowledge about the specific functions of GABAergic receptors in a neural loop is limited, there is evidence that suggests that the same subtype of GABAergic receptors can be expressed in different neuronal populations with different subcellular locations and in different areas of the brain, modulating the excitability and neuronal synchronization in different ways (Fig. 6). Therefore, understanding the role that GABA plays in the brain is possible only through the specific study of a receptor subtype, considering important variables such as synaptic activity, sex, or the circadian cycle, are partially explored along with GABA.
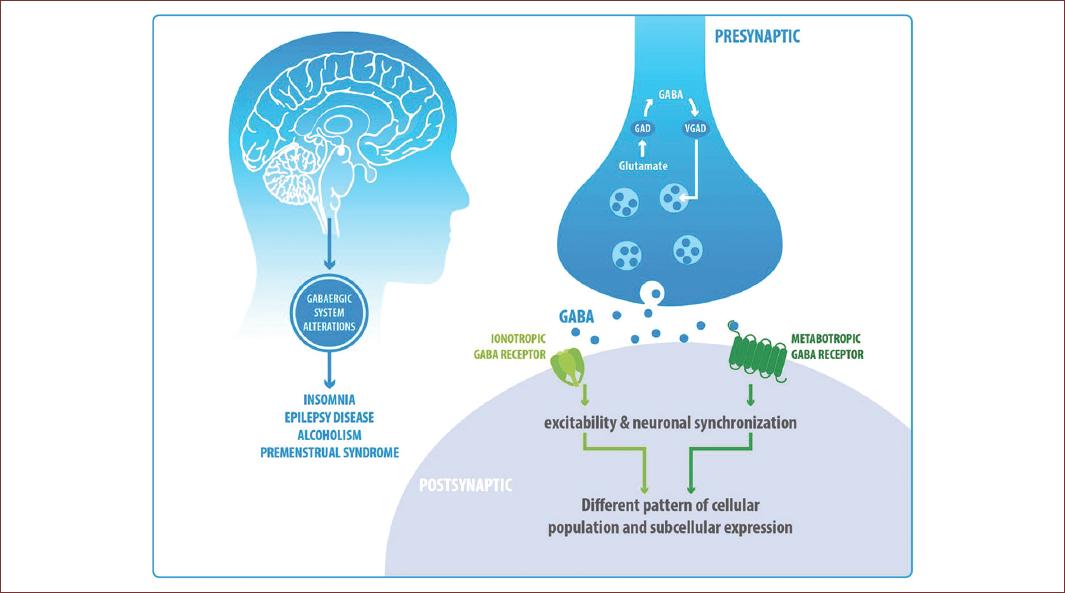
Figure 6 Its complex structural diversity, associated with its differential expression pattern in the neural loops of the brain, makes it very difficult to understand its inhibitory control in different neural networks. Although our knowledge about the specific functions of GABAergic receptors in a neural loop is limited, there is evidence that suggests that the same subtype of GABAergic receptors can be expressed in different neuronal populations with different subcellular locations and in different areas of the brain, modulating the excitability and neuronal synchronization in different ways (Fig. 6). Therefore, understanding the role that GABA plays in the brain is possible only through the specific study of a receptor subtype, a loop, or a neuronal type, considering important variables such as previous synaptic physiology, synaptic activity, sex, or the circadian cycle, are partially explored along with GABA.

