











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Viral, bacterial, and parasitic diseases have profoundly harmed the health of millions of people at different times in history. Such diseases include Black Death (yersinia pestis), cholera (vibrio cholerae), malaria (plasmodium), and smallpox. More recently, HIV, dengue and coronavirus have appeared on the scene. However, fungal infections have rarely resulted in serious diseases, at least during the recorded history of human beings. However, there is something uniquely problematic about fungi. Unlike bacteria, they are eukaryotes and thus share many similarities with the cells of their human hosts. Whereas antibiotics only target prokaryotic cells, compounds that kill fungi also harm the eukaryotic host, which impairs the development of antifungal agents and makes these infections the most difficult to treat. Furthermore, fungal tropism is highly variable, as pathogens infect a wide range of cell types. Depending on the immunological status of the host, a single fungal pathogen may infect multiple tissues in the same patient [1].
Among the estimated 1.5-5 million fungal species on the planet, those able to cause disease in humans are only a few hundred. Of these, a small number fulfill the four basic conditions necessary to affect healthy people: high temperature tolerance, ability to invade the human host, lysis and absorption of human tissue, and resistance to the human immune system. It is unusual for fungal disease to take hold in healthy individuals because the immune system of humans (and animals) is sophisticated, having evolved in constant response to fungal challenges. In contrast, fungal diseases occur frequently in immunocompromised patients. The four major fungal phyla that infect humans are Entomophthoromycota (Conidiobolus spp. and Basidiobolus spp.), Ascomycota (e.g. Candida spp. Fusarium spp., Histoplasma spp., Aspergillus spp., Coccidioides spp., and Pneumocystis spp.), Basidiomycota (Cryptococcus spp. and Trichosporon spp.) and Mucorales (Mucor spp. and Rhizopus spp.). [2-3]
Due to the increasing rate of fungal infections in hospitalized and immunocompromised patients, there is an urgent need to discover new antimycotic drugs. The biological attributes of triazole scaffolds, including those of the 1,2,3- and 1,2,4-isomers, are well recognized in the field of medicinal chemistry [4,5,6,7,8,9,10]. Many FDA-approved drugs contain such cores, being more common those with the 1,2,3-isomer: tazobactam and cefatrizine (broad-spectrum antibacterial agents), rufinamide (an anticonvulsant), suvorexant (a medication for insomnia), ticagrelor (a treatment to prevent stroke, heart attack and other adverse events in people with acute coronary syndrome) and bisoctrizole (a broad-spectrum chromophore added to sunscreens) (Scheme 1). Regarding conventional chemotherapy treatments for invasive, mucosal, and superficial fungal infections, the field is dominated by the 1,2,4-isomer. Itraconazole, terconazole, fluconazole and posaconazole (among others) have been conventional 1,2,4-triazole fungicidal agents for over 30 years, while efinaconazole and isavuconazole were more recently approved by the FDA (in 2014 and 2015, respectively) [11,12,13,14,15,16,17].
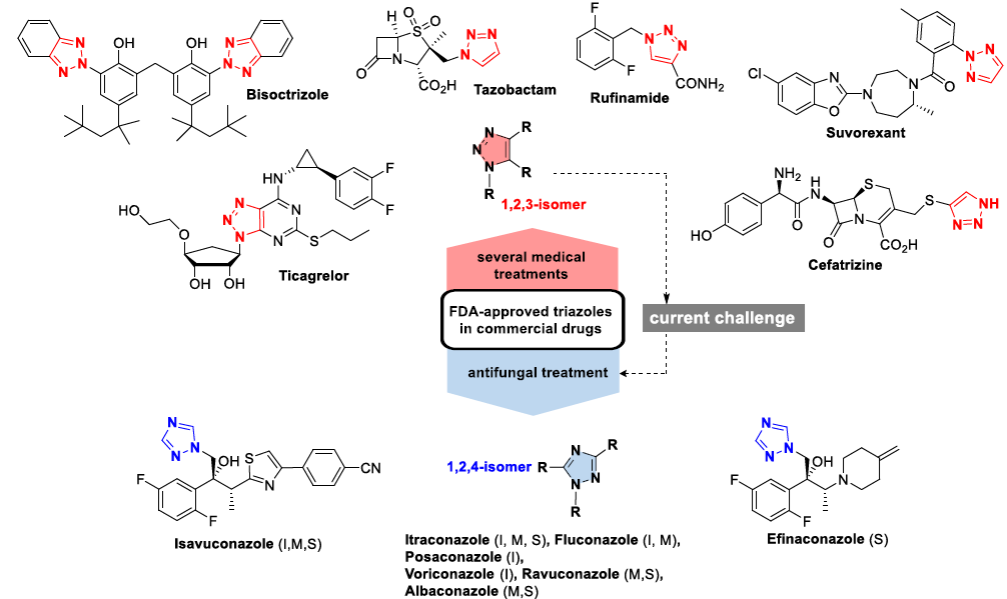
Scheme 1 Clinical drugs that are triazole derivatives, based on the 1,2,3- or 1,2,4-isomer. The1,2,4-isomer is part of the structure of some conventional pharmaceuticals used to treat invasive (I), mucosal (M), and superficial (S) fungal infections. The 1,2,3-triazole scaffold provides the basis for the antifungal drugs of choice.
For pathogenic fungi, as with all microorganisms, there is the specter of the emergence of strains resistance to pharmaceuticals [18,19,20,21], particularly those observed for Rhizopus (mucormycosis) as 1,2,4-triazole-drug resistant pathogen [22-23]. To meet this challenge, it is necessary to design and develop drugs that have well-defined and fungal-specific targets. The principal molecular target of azole antifungals is a protein, known as Erg11p or Cyp51p (according to distinct gene-based nomenclatures), in cytochrome P450. Cyp51p catalyzes the oxidative removal of the 14a-methyl group of lanosterol and/or eburicol in fungi by mono-oxygenase activity typical of P450. It contains an iron protoporphyrin moiety located at the active site. A bond is formed between a nitrogen atom in the triazolic ring and the iron atom in the protoporphyrin moiety. The remainder of the molecule apart from the triazole ring determines the manner in which a particular azole binds to the apoprotein and is also responsible for the wide variety of azole molecules [24,25,26,27]. Since the mechanism of action depends on the triazole moiety, researchers continue to seek new antifungal drugs with the triazole core, despite the mechanisms of resistance exhibited by some fungal pathogens.
The recent efforts by various research groups to elaborate 1,2,3-triazole isomers as antifungal drugs have yielded promising compounds with a good in vitro antifungal effect [28,29,30,31,32]. The ongoing research by our group is focused on the development of novel antifungal 1,2,3-triazoles (e.g., A, B and C, Scheme 2) [33,34,35] with dual pharmacophoric derivatives. Of the two linked pharmacophores, the main one is a 1,2,3-triazole core. Since the benzylic group has been recognized as an efficient pharmacophore [36,37,38,39], a novel library of benzylic 1,2,3-triazole derivatives was presently developed and tested. Related compounds are described in the literature as having efficient fungicidal activity (e.g., D and E) [40-41].

Experimental
Chemistry
All chemicals were of analytical grade and acquired from Merck and Sigma-Aldrich Company. Flash column chromatography was carried out by utilizing SiO2 60 (230-400 mesh). Reactions were monitored by TLC by using silica plates 60 and F254 aluminum sheets and were visualized with UV light at 254 nm. Melting points were determined for all newly synthesized compounds in open capillary tubes on a Fischer-Johns Scientific melting point apparatus. 1H and 13C NMR spectra were recorded on Bruker Avance 300 MHz and Varian 500 MHz instruments, with δ expressed in ppm and Me4Si as the internal standard.
General procedure for the synthesis of benzylic 1,2,3-triazole-4-carboxamides (3a-m)
To a solution of β-ketonitrile 2 (0.5 mmol) in t-BuOH anh. (1.0 mL) was added DBU (0.6 mmol) and benzyl azide 1 (0.5 mmol) under inert atmosphere. The reaction mixture was stirred at 70 ºC for 24 h. The progress of the reaction was monitored by TLC, which indicated when the starting materials disappeared. At this point, t-BuOK (1.5 mmol) was added to the reaction mixture, followed by continuous stirring for 10 h at room temperature. Upon completion of this time, TLC evidenced the appearance of the corresponding 1,2,3-triazole-4-carboxamide (3). Brine (~30 mL) was added and then the reaction mixture was washed with EtOAc (3×10 mL). The organic layer was dried with Na2SO4 and the solvent was evaporated under reduced pressure. Flash column chromatography furnished the pure triazole.
5-([1,1'-biphenyl]-4-yl)-1-(2,6-dichlorobenzyl)-1H-1,2,3-triazole-4-carboxamide (3a)
Following the general procedure, 3a was isolated as a yellowish solid (72%); mp 81-83°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.77 - 7.57 (m, 5H), 7.49 - 7.35 (m, 3H), 7.58 - 7.34 (m, 5H), 7.34 - 7.175 (m, 2H), 7.11 (s, 1H, NH), 5.65 (s, 2H, CH2); 13 C-NMR (75 MHz, CDCl3): δ (ppm) 162.30 (C=O), 142.86, 140.19, 139.78, 137.85, 136.70, 130.69, 130.39, 129.84, 129.10, 128.93, 128.53, 127.89, 127.70, 127.30, 127.22, 124.60, 48.12 (CH2).; EIMS m/z 422 (23), 221 (11), 159 (36), 43 (100).
5-([1,1'-biphenyl]-4-yl)-1-(3,4-bis(benzyloxy)benzyl)-1H-1,2,3-triazole-4-carboxamide (3b)
Following the general procedure, 3b was provided as white crystals (69%); mp 141-143°C; Rf: 0.16 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.64 - 7.56 (m, 4H), 7.50 - 7.21 (m, 15H), 7.11 (s, 1H, NH), 6.78 (d, J = 8.2 Hz, 1H), 6.59 (d, J = 2.1 Hz, 1H), 6.54 (dd, J = 8.2, 2.1 Hz, 1H), 5.48 (s, 1H, NH), 5.35 (s, 2H, CH2), 5.12 (s, 2H, -O-CH2), 5.01 (s, 2H, -O-CH2).; 13 C-NMR (75 MHz, CDCl3) δ (ppm): 162.03(C=O), 148.92, 142.74, 140.03, 139.31, 138.45, 136.95, 136.73, 130.40, 128.94, 128.51, 128.49, 127.93, 127.89, 127.72, 127.22, 127.19, 127.15, 124.62, 120.84, 114.85, 114.24, 71.15(O-CH2), 71.01 (O-CH2), 51.94 (CH2).; EIMS m/z 566 (10), 355 (8), 212 (10), 91 (100).
5-([1,1'-biphenyl]-4-yl)-1-(2,3-dimethoxybenzyl)-1H-1,2,3-triazole-4-carboxamide (3c)
Following the general procedure, 3c was afforded as yellowish crystals (68%); mp 142-145°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.67 - 7.57 (m, 4H), 7.50 - 7.33 (m, 5H), 6.97 (t, J = 8.0 Hz, 1H), 6.86 (dd, J = 8.0, 1.6 Hz, 1H), 6.47 (dd, J = 7.9, 1.5 Hz, 1H), 5.65 (s, 1H, NH), 5.51 (s, 2H, CH2), 3.83 (s, 3H, OMe), 3.61 (s, 3H, OMe).; 13 C-NMR (75 MHz, CDCl3): δ (ppm) 162.39 (C=O), 152.58, 146.31, 142.80, 140.22, 139.80, 138.22, 137.88, 130.48, 129.51, 129.05, 128.24, 127.86, 127.23, 125.32, 124.65, 124.26, 120.13, 112.72, 60.41(OMe), 55.78 (OMe), 47.27(CH2).; EIMS m/z 414 (80), 221 (8), 151 (50), 136 (90), 91 (100).
1-(2,6-dichlorobenzyl)-5-phenyl-1H-1,2,3-triazole-4-carboxamide (3d)
The general procedure gave 3d as a yellowish powder (65%); mp 200-201°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.59 - 7.38 (m, 5H), 7.33 - 7.17 (m, 3H), 7.07 (s, 1H, NH), 5.62 (s, 1H, NH), 5.59 (s, 2H).; 13 C-NMR (75 MHz, CDCl3): δ (ppm) 160.32 (C=O), 138.10, 136.00, 134.86, 128.85, 128.22, 128.12, 127.99, 126.75, 126.68, 124.02, 46.15 (CH2).; EIMS m/z 346 (10), 311 (34), 187 (3), 159 (100), 89 (48).
1-(benzo[d][1,3]dioxol-5-ylmethyl)-5-phenyl-1H-1,2,3-triazole-4-carboxamide (3e)
Following the general procedure, 3e was produced as a white powder (75%); mp 170-173°C; Rf: 0.14 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, DMSO-d6): δ (ppm) 7.83 (s, 1H, NH), 7.62 - 7.40 (m, 3H), 7.43 - 7.32 (m, 2H), 6.78 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 1.7 Hz, 1H), 6.37 (dd, J = 8.0, 1.8 Hz, 1H), 5.97 (s, 2H, -OCH2O-), 5.39 (s, 2H, CH2).; 13 C-NMR (75 MHz, DMSO-d6): δ (ppm) 162.24 (C=O), 147.86, 147.41, 139.57, 138.90, 130.47, 130.01, 129.36, 128.76, 126.67, 121.43, 108.71, 108.19, 101.64(-OCH2O-), 51.54 (CH2).; EIMS m/z 322 (25), 293 (25), 149 (20), 135 (100), 77 (60).
1-(3,4-bis(benzyloxy)benzyl)-5-phenyl-1H-1,2,3-triazole-4-carboxamide (3f)
Following the general procedure, 3f was furnished as a yellowish powder (69%); mp 111-114°C; Rf: 0.14 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, DMSO-d6): δ (ppm) 7.84 (s, 1H, NH), 7.55 - 7.25 (m, 15H), 6.93 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 2.0 Hz, 1H), 6.47 (dd, J = 8.3, 2.0 Hz, 1H), 5.38 (s, 2H, CH2), 5.07 (s, 2H, -O-CH2), 4.93 (s, 2H, -O-CH2).; 13 C-NMR (75 MHz, DMSO-d6): δ (ppm) 161.85 (C=O), 148.07, 147.99, 139.15, 138.44, 137.12, 136.93, 130.08, 129.53, 128.46, 128.43, 128.32, 128.09, 127.90, 127.85, 127.68, 127.56, 126.37, 120.37, 114.40, 113.73, 70.07 (O-CH2), 51.12 (CH2).
1-(2,3-dimethoxybenzyl)-5-phenyl-1H-1,2,3-triazole-4-carboxamide (3g)
The general procedure led to 3g as a white powder (71%); mp 206-208°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, DMSO-d6): δ (ppm) 7.66 (s, 1H, NH), 7.52 - 7.34 (m, 5H), 7.32 (s, 1H, NH), 7.02 - 6.90 (m, 2H), 6.39 (dd, J = 5.5, 3.7 Hz, 1H), 5.42 (s, 2H, CH2), 3.79 (s, 3H, OMe), 3.46 (s, 3H, OMe).; 13 C-NMR (125 MHz,CDCl3/DMSO-d6): δ (ppm) 162.19 (C=O), 152.54, 146.24, 139.12, 130.36, 129.74, 128.99, 128.49, 126.69, 124.30, 120.37, 113.15, 60.00 (OMe), 55.98 (OMe), 47.07 (CH2).; EIMS m/z 338 (75), 307 (62), 136 (91), 91 (100).
1-(4-methoxybenzyl)-5-phenyl-1H-1,2,3-triazole-4-carboxamide (3h)
The general procedure resulted in 3h as a white powder (70%); mp 209-210°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.56 - 7.38 (m, 3H), 7.34 - 7.23 (m, 2H), 7.08 (s, 1H, NH), 6.95 (d, J = 8.2 Hz, 2H), 6.78 (d, J = 8.1 Hz, 2H), 5.47 (s, 1H, NH), 5.36 (s, 2H, CH2), 3.77 (s, 3H, OMe).; 13 C-NMR (125 MHz, CDCl3): δ (ppm) 162.01 (C=O), 159.60, 139.49, 138.38, 129.97, 129.08, 128.55, 128.50, 126.69, 125.90, 114.15, 55.27 (OMe), 51.65 (CH2).; EIMS m/z 308 (60), 279 (55), 121 (100), 77 (40).
1-(2,6-dichlorobenzyl)-5-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3i)
Following the general procedure, 3i was obtained as yellowish crystals; yield (76%); mp 187-189°C; Rf: 0.14 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.38 (d, J = 8.2 Hz, 2H), 7.34 - 7.25 (m, 4H), 7.22 (dd, J = 9.2, 6.7 Hz, 1H), 7.07 (s, 1H, NH), 5.69 (s, 1H, NH), 5.57 (s, 2H, CH2), 2.42 (s, 3H, CH3).; 13 C-NMR (75 MHz, CDCl3): δ (ppm) δ 162.29 (C=O), 140.16, 139.99, 137.67, 136.64, 130.58, 129.90, 129.79, 129.29, 128.43, 122.72, 47.80 (CH2), 21.44 (CH3).; EIMS m/z 361 (19), 325 (30), 159 (100), 77 (21).
1-(benzo[d][1,3]dioxol-5-ylmethyl)-5-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3j)
Following the general procedure, 3j was formed as yellow crystals (76%); mp 157-159°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.26 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.2 Hz, 2H), 7.08 (s, 1H, NH), 6.68 (d, J = 8.0 Hz, 1H), 6.57 (d, J = 1.8 Hz, 1H), 6.48 (dd, J = 8.0, 1.8 Hz, 1H), 5.93 (s, 2H, -OCH2O-), 5.64 (s, 1H, NH), 5.31 (s, 2H, CH2), 2.42 (s, 3H, CH3). 13 C-NMR (125 MHz, CDCl3): δ (ppm) 162.16 (C=O), 147.99, 147.65, 140.19, 139.63, 138.28, 129.79, 129.30, 128.42, 122.66, 121.32, 108.29, 108.11, 101.25, 51.70 (-OCH2O-), 21.43 (CH3).; EIMS m/z 336 (32), 149 (12), 135 (100), 77 (56).
1-(3,4-bis(benzyloxy)benzyl)-5-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3k)
Following the general procedure, 3k was prepared as colorless crystals (73%); mp 117-119°C; Rf: 0.16 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) δ 7.46 - 7.24 (m, 10H), 7.19 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 8.2 Hz, 3H, Ar, NH,), 6.78 (d, J = 8.2 Hz, 1H), 6.59 (d, J = 2.1 Hz, 1H), 6.53 (dd, J = 8.2, 2.1 Hz, 1H), 5.52 (s, 1H, NH), 5.29 (s, 2H, CH2), 5.12 (s, 2H, -OCH2), 5.02 (s, 2H, -OCH2), 2.39 (s, 3H, CH3).; 13 C-NMR (125 MHz, CDCl3): δ (ppm) 162.09 (C=O), 148.89, 148.86, 140.06, 139.59, 138.27, 136.95, 136.79, 129.79, 129.24, 128.49, 128.48, 127.90, 127.85, 127.23, 127.20, 122.73, 120.80, 114.84, 114.27, 71.15 (-OCH2), 71.03 (-OCH2), 51.69 (CH2), 21.45 (CH3).; EIMS m/z 504 (12), 413 (2), 293 (11), 91 (100).
1-(2,3-dimethoxybenzyl)-5-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3l)
The general procedure gave 3l as a white powder; yield (82%); mp 185-188°C; Rf: 0.14 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.24 (s, 4H), 7.12 (s, 1H, NH), 6.97 (t, J = 8.0 Hz, 1H), 6.88 (td, J = 8.1, 1.6 Hz, 1H), 6.43 (dd, J = 8.1, 1.6 Hz, 1H), 5.66 (s, 1H, NH), 5.46 (s, 2H, CH2), 3.84 (s, 3H, OMe), 3.62 (s, 3H, OMe), 2.38 (s, 3H, CH3).; 13 C-NMR (75 MHz, CDCl3): δ (ppm) 162.29 (C=O), 152.45, 146.14, 140.02, 140.00, 138.04, 129.77, 129.20, 128.83, 124.17, 122.67, 119.95, 112.52, 60.20 (OMe) , 55.71 (OMe), 46.96 (CH2), 21.37 (CH3).; EIMS m/z 352 (55), 321 (58), 136 (100), 92 (81).
1-(4-methoxybenzyl)-5-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3m)
The general procedure produced 3m as a white powder; yield (69%); mp 190-191°C; Rf: 0.15 (Hex/EtOAc 70/30 X3). 1 H-NMR (300 MHz, CDCl3): δ (ppm) 7.26 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H), 7.07 (s, 1H, NH), 7.02 - 6.96 (m, 2H), 6.82 - 6.76 (m, 2H), 5.56 (s, 1H, NH), 5.35 (s, 2H, CH2), 3.77 (s, 3H, OMe), 2.42 (s, 3H, CH3).13 C-NMR (75 MHz, CDCl3): δ (ppm) 162.15 (C=O), 159.56, 140.12, 139.60, 138.27, 129.83, 129.28, 129.04, 126.87, 122.76, 114.12, 55.25 (OMe), 51.47 (CH2), 21.46 (CH3).; EIMS m/z 322 (25), 136 (35), 121 (100), 91 (40).
Fungicidal activity
The antifungal impact of 3a-3m was analyzed in vitro with the microdilution techniques for testing antimicrobial susceptibility. All assays were performed in triplicate. The M38-A2 method was employed to examine activity against filamentous fungi [42,43,44], while the M27-A3 method described by the CLSI was adopted to assess the effects against yeasts [45,46,47].
The filamentous fungal strains consisted of M. hiemalis ATCC-8690, A. fumigatus ATCC-16907, T. cutaneum ATCC-28592 and R. oryzae ATCC-10329. The yeast specimens were comprised of C. albicans ATCC-10231, C. utilis ATCC-9226, C. krusei ATCC-14243 and C. glabrata ATCC-34138.
For antifungal evaluations, strains of clinical importance in Mexico, Latin America and Europe were considered. Candidiasis is the most frequent superficial and systemic mycosis in neonates, immunocompromised individuals, and hospitalized patients [48]. One strain of filamentous fungi from each of the genera Rhizopus, Aspergillus and Mucor can cause systemic infections and severe gastrointestinal disorders [49]. The fungi were cultured in RPMI 1640 synthetic medium containing glutamine but not sodium bicarbonate, a morpholino propane sulfonic acid (MOPS) buffer at 0.164 M adjusted to pH 7±0.1, and 0.2% glucose.
Because the synthesized compounds are not soluble in water, itraconazole served as the standard drug in the exploration of antimicrobial sensitivity. Based on the CLSI methodology that establishes the microdilutions for the reference drug, a solution of itraconazole at 1600 μg/ml was dissolved in dimethyl sulfoxide to reach concentrations ranging from 16 µg/mL to 0.03 µg/mL, and the corresponding values in mmol/mL were determined and utilized for the solutions of 3a-3m (Table 1).
Table 1 Concentrations of itraconazole used to assess antifungal sensitivity, following the protocol of the CLSI.
| CLSI µg/mL | 16 | 8 | 4 | 2 | 1 | 0.5 | 0.25 | 0.12 | 0.06 | 0.03 |
| µmol/mL | 2.6 | 1.13 | 0.56 | 0.28 | 0.14 | 0.07 | 0.035 | 0.017 | 0.008 | 0.004 |
In accordance with the CLSI, the minimum inhibitory concentrations (MIC) of the compounds and the standard drug were determined with the help of an inverted mirror, performing all experiments in triplicate. The different concentrations of the test compounds and reference drug were added to 96-well plates containing RPMI 1640 medium buffered with MOPS (3-[N-morpholino] propane sulfonic acid) (Sigma-Aldrich). MIC values are expressed in mmol per milliliter.
After 24 h growth on SDA plates, 3-5 fungal colonies ≥1 mm were gathered with a culture loop, resuspended in saline solution (0.85 % NaCl), stirred well and adjusted to 0.5 McFarland optical density with the aid of a spectrophotometer (wavelength 530 nm). This solution, having a concentration of 1-5 x 106 CFU/ml, was diluted (1:1000) with RPMI medium and utilized to inoculate the plates containing the antifungal compounds. The wells of columns 2-11 were inoculated with 100 μl of the yeast suspension. Column 1 had 200 μl of RPMI in each well and did not receive inoculation, thus serving as the sterility control of the medium. The wells in column 12 were inoculated but did not contain any antifungal compound, serving as the growth control. For all strains of yeast and filamentous fungi, the plates were incubated at 35 °C for 48 h.
The visual reading of fungal growth was carried out with an inverted mirror. For the azoles, the MIC is the lowest concentration of an antifungal that produces a substantial reduction in yeast growth (≥50%) and a 100% inhibition of the growth of filamentous fungi, in each case compared to the corresponding control.
Results and discussion
Chemistry
Our group recently reported a novel two-step methodology for synthesizing benzylic 1,2,3-triazole-4-carboxamide derivatives 3 [50]. The first step is a highly regioselective azide-enolate 1,3-dipolar cycloaddition (coupling benzyl azide 1/β-ketonitrile 2) in dimethylformamide to generate the benzylic 1,2,3-triazole-4-carbonitrile adducts 4, followed by their isolation and identification. The second step is a hydrolysis of the nitrile group to afford triazolic compounds 3 as antifungal agents.
A library of novel benzylic 1,2,3-triazole-4-carboxamide derivatives was presently designed and then elaborated via an optimized protocol involving a one-pot reaction that facilitates the in situ hydrolysis of the nitrile (Scheme 3). Thirteen novel benzylic 1,2,3-triazole-4-carboxamides (3a-m) were obtained with acceptable yields (65-82%). For the current synthetic procedure, the solvent was tert-butanol rather than dimethylformamide. The latter, included in the previous two-step protocol, had the drawback of remaining in the reaction products during the purification process. Hence, the one-step methodology is eco-friendly. For the 1,3-dipolar cycloaddition (promoted by DBU), the temperature was 70 ºC and the reaction time 24 h. For the hydrolysis, the second part of the one-pot reaction was carried out at room temperature for 10 h. Anhydrous t-BuOH proved to be an excellent solvent.
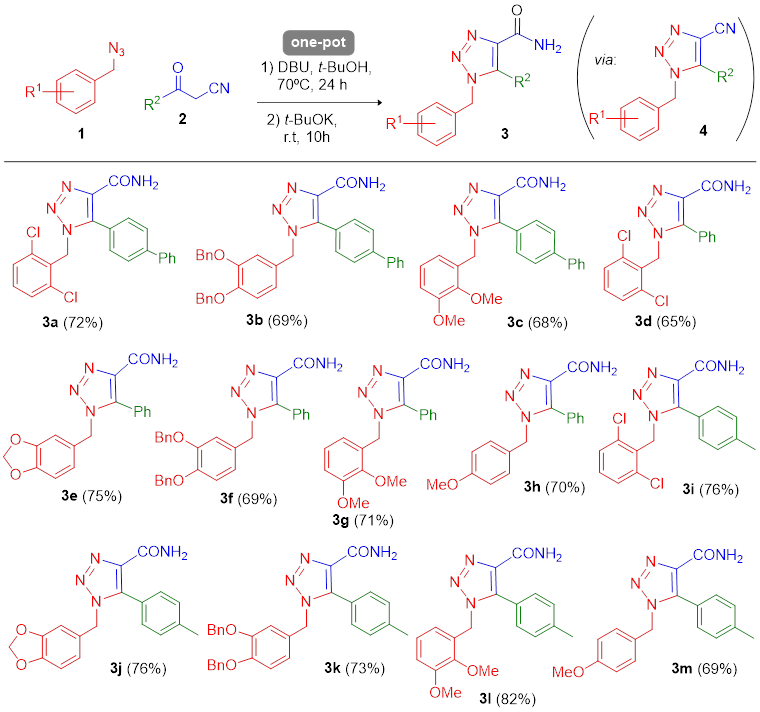
Scheme 3 One-pot synthesis of 1,2,3-triazole-4-carboxamides 3a-m via hydrolysis of 5-nitrile substituted triazolic intermediates (4). The latter were afforded by azide-enolate cycloaddition (pericyclic coupling between azides 1 and β-ketonitriles 2). Reaction conditions: β-ketonitrile 2 (1.0 eq), benzyl azide 1 (1.0 eq), and DBU (1.1 eq) in anh. t-BuOH under inert atmosphere was stirred at 70 ºC for 24 h. Then t-BuOK (3.0 eq) was added, and the reaction mixture was stirred at rt for 10 h.
Fungicidal activity
According to the microdilution techniques of antifungal susceptibility testing protocols (CLSI), itraconazole was prepared at ten concentrations from 16 µg/mL to 0.03 µg/mL, as shown in Table 1. The corresponding values in mmol/mL were determined and utilized for the solutions of 3a-3m.
The outcome of the in vitro antifungal evaluation of 3a-3m and itraconazole is expressed as the mean of the MIC of three assays (Table 2), found in relation to the filamentous fungi (M. hiemalis ATCC-8690, A. fumigatus ATCC-16907, T. cutaneum ATCC-28592 and R. oryzae ATCC-10329) and the yeasts (C. albicans ATCC-10231, C. utilis ATCC-9226, C. krusei ATCC-14243 and C. glabrata ATCC-34138).
Table 2 The in vitro antifungal activity of the synthesized compounds is expressed as the MIC (µmol/mL).
| Yeasts | Filamentous fungi | ||||||||
| Compound | C. alb. | C. uti. | C. kru. | C. gla. | M. hie. | A. fum. | T. cut. | R. ory. | |
| 3a | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 0.14 | |
| 3b | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 0.14 | |
| 3c | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 0.07 | |
| 3d | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 0.017 | |
| 3e | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 0.017 | |
| 3f | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | |
| 3g | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | |
| 3h | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 1.13 | |
| 3i | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 1.13 | 2.6 | 1.13 | |
| 3j | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 1.13 | 2.6 | 2.6 | |
| 3k | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | |
| 3l | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | |
| 3m | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | 2.6 | |
| Standard a | 0.004 | 0.035 | 0.035 | 0.14 | 0.56 | 0.14 | 1.13 | 0.14 | |
Abbreviations: C. alb., Candida albicans; C. uti., Candida utilis; C. kru., Candida krusei; C. gla., Candida glabrata; M. hie, Mucor hiemalis; A. fum, Aspergillus fumigatus; T. cut, Trichosporon cutaneum; R. ory, Rhizopus oryzae; a Itraconazole.
Previous studies have demonstrated that certain triazoles exhibit in vitro activity against several human pathogenic fungi, including Candida species and filamentous fungi such as Aspergillus spp., Mucor spp. and Rhizopus spp. [33]. In the current contribution, thirteen synthesized compounds were tested in vitro against four filamentous fungi and four yeast specimens.
The inhibition of R. oryzae was excellent for 3d and 3e and good for 3c, in the three cases better than the results obtained with the reference drug, itraconazole (3d and 3e, MIC = 0.017 µmol/mL; 3c, MIC = 0.07 µmol/mL; itraconazole, MIC = 0.14 µmol/L). Compounds 3a and 3b displayed an antifungal effect on R. oryzae equivalent to that of itraconazole.
None of the test compounds were active against the yeast strains or two of the filamentous fungi, M. hiemalis and T. cutaneum. The treatment with 3i and 3j led to a very limited impact on A. fumigatus. No structure-activity correlation was found for the yeast growth inhibition of C. albicans induced by the active compounds.
For both 3d and 3e (the most efficient fungicidal agents against R. oryzae), the structure-activity relation pointed to the 4-phenyl-4-carboxamide triazole moiety as being responsible for the antifungal effect, while neither the electron-poor ring (substituted with 2,6-dichloro to form 3d) or electron-rich ring (containing piperonyl in 3e) were relevant. Small ethers as substituents improved the results obtained, as evidenced by the data for 3c and 3e. Another critical aspect is the size of the structure. 3a, 3d and 3e are the smallest of the series of synthesized compounds and constitute three of the five molecules with the lowest MIC values for activity against R. oryzae.
The compounds that showed less activity than the reference drug bear the largest functional groups in their structure, suggesting the importance of a small substituent in positions 1 and 5 of the triazole to favor interaction with the active site of the fungus.
The antifungal effect of 3d and 3e in R. oryzae is of great value because this fungus produces infections in immunocompetent patients that can lead to mucormycosis [51-52]. Furthermore, since R. oryzae had showed resistance against 1,2,4-triazolic isomer commercial drugs [22-23], these outcomes represent an excellent opportunity for 1,2,3-triazolic isomers as serious candidates in the treatment of such infections.
Conclusion
Thirteen benzylic 1,2,3-triazole-4-carboxamides 3a-m were elaborated by a novel one-pot procedure, avoiding previous purifications and the expenditure of organic solvents. Thus, the synthetic protocol is eco-friendly. In addition, the raw materials for the reaction are economical and allow for an easy synthesis in the lab.
Based on in vitro microdilution techniques for the evaluation of antimicrobial susceptibility, 3d and 3e proved to be the most efficient fungicidal agents against R. oryzae, showing greater antifungal activity than the other test compounds and the reference drug, itraconazole. Since R. oryzae had showed resistance against 1,2,4-triazolic isomer commercial drugs, 3d and 3e are 1,2,3-triazolic candidates for future complementary biological studies in order to increase the antifungal effect by optimizing these two new scaffolds. Future research could possibly involve the optimized compounds in clinical trials to treat clinical infections produced by Rhizopus spp. strains, especially mucormycosis in immunocompetent patients.

