











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The COVID-19 pandemic continues today with almost 575 million infections and 6.4 million deaths (July 30, 2022) [1], and at the end of 2021, variants of SARS-CoV-2 with different characteristics were identified that have contributed to new waves of infections and to the delay of this pandemic [2-4]. Therefore, several therapeutic targets are being proposed against SARS-CoV-2, that interacting with ACE2, it protein plays a really important to develop the SARS-CoV-2 infectious process. Lastly, in the SARS-CoV-2 variants have been reported mutations in the receptor-binding domain (RBD) that could increase the infectious process and decreases the effect of vaccines, in which it is reported that the mutation in the position 452, 478, 484 and 501 in SARS-CoV-2 increase the transmissibility and infectivity of this virus, since it favors a greater interaction of RBD with ACE2 [5,6], and it is theoretically demonstrated too, that some mutations can improve the interaction between RBD and ACE2 (N501Y, D614G, A845V, and P1143L), it shows greater interaction with the ACE2 receptor than native [5,7].
Nowadays, there have been proposed different therapeutic targets to develop drugs along this pandemic, SARS-CoV-2 proteases (3CLpro and PLpro), RNA-Dependent RNA Polymerase (RdRp) [8-10], membrane fusion inhibitors (HR1 and HR2 of S-protein) [11-14], Spike-Protein (S-protein) of SARS-CoV-2 [15, 16], and the continuous development of vaccines and their implementation [17-19], but lastly all these developments affected by SARS-CoV-2 variants. Given the presence of new mutations in this virus, the need to develop specific drugs against this disease continues, and this study proposes a potential characteristic site in the SARS-CoV-2 variants that have emerged by the end of 2021 [20, 21], so this study starts from the region of interaction between the RBD of the S-Protein SARS-CoV-2 with the ACE2 [22, 23], which has been used for several in silico works, performing molecular docking against RBD wild type (WT) close to amino acid 501 [24-26], RBD-WT (Phe486, Asn487, Tyr489) [27], and from compounds targeted between amino acids 402 to 505 in the RBD [28]. But there are no studies to date focused on this characteristic region in SARS-CoV-2 variants (between amino acids Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and, Tyr489), in which the SARS-CoV-2 variants of concern have mutations in this region (Beta, Gamma, Delta, Kappa, Eta and Omicron) [6], so this site could contribute to the interaction between the S-protein and ACE2.
This study proposes compounds selected by molecular docking directed to a specific potential site in the RBD of SARS-CoV-2 variants. We used the crystallographic structure with the amino acid mutations in the SARS-CoV-2 variants (in position 478 and 484), this site is important for interaction between SARS-CoV-2 (RBD) and ACE2 [23,29]; to select the compounds with better average of binding that could difficult/block the interaction between S-protein and ACE2 to develop a new way against COVID-19. Will are proposing compounds with a high potential to be inhibitors for SARS-CoV-2 variants and to contributing in the efforts to develop a new drug.
Experimental
Materials and methods
The build model of RBD of SARS-CoV-2 variants
We used the atomic coordinates of the Receptor-Binding Domain (RBD) (PDB:6M17) to build a model of RBD-SARS-CoV-2 variant (PDB-variant) for molecular docking [30] using the SWISS-MODEL server (build model) [31]. The structure (PDB:6M17) was modified in the positions 478 and 484 (T478K and E484K), to emulate the mutations in the SARS-CoV-2 variants (Beta, Gamma, Delta, Kappa, Eta and Omicron) [6], and the built model (PDB-variant) was validated on RAMPAGE and SAVES 6.0 web servers [32].
Preparation of receptor protein and definition of binding site
We used the atomic coordinates of the model generated for the RBD in SARS-CoV-2 variants. The protonation and energy minimization of PDB file was performed using Molecular Operating Environment (MOE), with the default parameters and the CHARMM27 force field [33,34], and the specific binding site for molecular docking procedures are the amino acids: Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and Tyr489 in the RBD.
Compound library
We used the EXPRESS-pick Collection Stock of the small molecule screening library from Chembridge Corp. for molecular docking [35]. This collection of small molecule screening compounds contains over 500,000 chemical compounds that fulfill the druggable properties of Lipinski’s rules [36,37] and cover a broad area of chemical space.
Molecular docking
The molecular docking was carried out following the reported methodology [35,38-40] by MOE software, using default parameters (Placement: Triangle Matcher, Rescoring 1: London ΔG, Refinement: Forcefield, Rescoring 2: London ΔG), for each compound were generated up to 100 conformers.
Calculation of the free binding energy (ΔGbinding)
The binding affinity of each complex (Ligand-Protein) was estimated by the ratio of General Born vs Volume Integral (GB/VI) [41], using parameters in MOE [42,43]. General Born or non-bonded interaction energies comprise Van der Waals, Coulomb electrostatic interactions and implied solvent interaction energies [43].
Selection of the best ten compounds
To select the best ten compounds, each compound was simulated with up to 100 conformations in the docking, with these results were analyzed the best 30 conformers of each compound to determine their averages of ΔGbinding with Excel (Microsoft) following the reported methodology [38,39], and the best ten compounds averages of ΔGbinding were determined for RBD-variant (mutations in positions 478 and 484). The description of chemical properties by PhysChem - ACD/Labs [44], the theoretical toxicity, carcinogenicity and mutagenicity were considered [45]. The calculated interactions between RBD and the compounds were visualized with Ligand-interaction interactions in MOE.
Results
Selection of compounds by docking
From docking´s results, by the interaction of more of 500,000 compounds in the RBD of SARS-CoV-2 (amino acids: Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and Tyr489, Fig. 1), we selected the best ten compounds (labeled V1 - V10), the selection criteria these compounds, was based on the calculation of the average of the ΔGbinding of each compound, using the ΔGbinding values of 30 conformers. We determined a range between -11.93 to -13.19 kcal/mol for the best ten compounds (Table 1, and details on the supplementary material in Table S1). These ten compounds (V1 - V10) are in the Express-pick Collection Stock of the small molecule screening of Chembridge library (ChemBridge Corp.) and the analysis of the interaction of each compound with RBD was carried out with the interaction report in MOE (Table 2 and details in Table S2 - S11). In addition, the description of the theoretical toxicity (Table S12), ADME characteristics (Table S13) and chemical properties of each compound (V1 - V10, Table S14), are presented in the supplementary material.
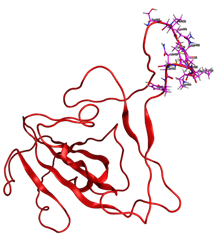
Fig. 1 RBD (Red) shows amino acids Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and Tyr489 (Pink), as the specific region chosen for molecular docking.
Table 1 PubChem CID, ID Chembridge compound and chemical structure the ten compounds, V1 - V10.
V1.- 2954504, 7786456
|
V2.- 2210833, 7827231 
|
V3.- 3808110, 7790487
|
V4.- 2211666, 7835824 
|
V5.- 909537, 6488828
|
V6.- 2954633, 7787498 
|
V7.- 2208327, 7804309
|
V8. - 2208995, 7808994 
|
V9.- 2205250, 7786334
|
V10.- 1296144, 7792008 
|
Table 2 PubChem CID and ID Chembridge compound, Canonical Smiles, Interaction with residues in RBD, Number of conformers, Average ΔGbinding (kcal/mol) with standard deviation (SD), Ames test and strain used (positive or negative) [57].
| PubChem CID, ID Chembridge compound | Canonical Smiles | Interaction with residues in RBD (Table S2 - S11) | Number of conformers | Average of ΔGbinding and SD (Table S1) | PreADMET Ames test -TA100_10RL -TA100_NA -TA1535_10R -TA1535_NA |
|---|---|---|---|---|---|
| 2954504, 7786456 | C1=COC(=C1)CNC(=S)NNC(=S)NC2=CC=C(C=C2)Cl | Gln474, Lys478, Cys480, Gly485, Phe486, Asn487, Cys488 |
30 | -13.19 2.59 | Mutagen -Negative -Positive -Positive -Negative |
| 2210833, 7827231 | COC1=C(C=C(C=C1)Cl)NC(=S)NNC(=S)NC2=CC=C(C=C2)Cl | Lys478, Pro479, Cys480, Gly485, Phe486, Asn487, Cys488 |
30 | -12.82 2.89 | Mutagen -Negative -Negative -Negative -Negative |
| 3808110, 7790487 | COC1=C(C=CC(=C1)[N+](=O)[O-])NC(=S)NNC(=S)NC2=C(C=C(C=C2)F)F | Lys478, Cys480, Gly482, Gly485, Phe486, Cys488 |
30 | -12.68 2.62 | Mutagen -Positive -Negative -Negative -Negative |
| 2211666, 7835824 | C1=CC=C2C(=C1)C=CC=C2NC(=S)NNC(=S)NC3=CC=C(C=C3)Cl | Lys478, Pro479, Cys480, Asn481, Lys484, Gly485, Phe486, Asn487, Cys488 |
30 | -12.64 4.11 | Non-mutagen -Negative -Negative -Negative -Negative |
| 909537, 6488828 | COC(=O)NNC(=O)NC1=CC=C(C=C1)Cl | Gln474, Lys478, Cys480, Lys484, Phe486, Asn487, Cys488 |
30 | -12.38 1.28 | Mutagen -Negative -Negative -Negative -Negative |
| 2954633, 7787498 | CCOC(=O)NNC(=O)NC1=CC=C(C=C1)Br | Lys478, Cys480, Gly485, Phe486, Asn487, Cys488 |
30 | -12.34 1.46 | Mutagen -Positive -Negative -Negative -Negative |
| 2208327, 7804309 | CC(C)(C)OC(=O)NNC(=S)NC1=C(C=C(C=C1)Br)Cl | Lys478, Cys480, Asn481, Gly485, Phe486, Cys488 |
30 | -12.28 2.37 | Mutagen -Positive -Positive -Negative -Negative |
| 2208995, 7808994 | COC(=O)C1=C(SC<2=C1CCC2)NC(=S)NNC(=S)NC3=C(C=C(C=C3)F)F | Lys478, Pro479, Cys480, Lys484, Gly485, Phe486, Cys488 |
30 | -12.04 1.85 | Mutagen -Negative -Positive -Negative -Negative |
| 2205250, 7786334 | CC(C)(C)OC(=O)NNC(=O)NC1=C(C(=CC=C1)Cl)Cl | Lys478, Cys480, Lys484, Gly485, Phe486, Asn487, Cys488 |
30 | -12.01 2.51 | Mutagen -Positive -Positive -Positive -Negative |
| 1296144, 7792008 | C1OC2=C(O1)C=C(C=C2)NC(=S)NNC(=S)NC3=C(C=C(C=C3)F)F | Lys478, Pro479, Cys480, Asn481, Lys484, Gly485, Phe486, Asn487, Cys488 |
30 | -11.93 2.11 | Mutagen -Negative -Negative -Negative -Negative |
Interaction of V1 - V10 compounds with RBD
For determining the probable interaction of each compound (V1 - V10) with RBD, we analyzed 30 conformers of each one. From docking result, we determined the main amino acids in RBD to interact with the ten compounds, the compounds are interacting mainly in the amino acids Gln474, Lys478, Pro479, Cys480, Asn481, Gly482, Lys484, Gly485, Phe486, Asn487, and Cys488 (Table 2, Table S2 - S11). The interaction of each compound and their conformers with RBD are shown in the supplementary material (Figures S1 - S10).
Discussion
The pandemic caused by the SARS-CoV-2 virus continues, as well as the development of drugs against this disease. It is important to develop different strategies against this virus, since SARS-CoV-2 variants have been presented that cause a decrease in the effect of vaccines and some drugs [7, 12-14, 6, 39, 46-49]. In this study was carried out a molecular docking aimed to the amino acids Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and Tyr489 in the RBD of SARS-CoV-2, these amino acids are important in SARS-CoV-2 variants to interact with ACE2 [50, 23 ,29], due to It is reported that some amino acids in this region, such as Asp480 and Phe486 destabilizes the interaction with ACE2, as well as the mutation in Ser494, Gln498 and Asn501 [51]. Therefore, this region (477-488 in RBD) could be a site to destabilize the interaction between S-protein and ACE2.
From docking results we determined the main amino acids for the ten compounds proposed, these are Gln474, Lys478, Pro479, Cys480, Asn481, Lys484, Gly485, Phe486, Asn487, and Cys488 in the RBD (Table 2), as we mention these amino acids have been reported for the interaction between SARS-CoV-2 with ACE2 [22, 23, 29, 52], and with the docking results we determined several interactions of hydrogen bridges of each compound proposed in the region where are the amino acids Lys 478 and Lys484 (Table S2-S11), and it is important to emphasize that blocking Lys 478 and Lys484 could decreases the interaction with ACE2. The mutations in the position 478 and 484 in the S-protein (Lys and Lys/Gln respectively) are proposed as a factor that could increase the interaction of RBD with ACE2 [50] [23] [29]. Therefore, this site of interaction in RBD could be as a therapeutic target to inhibit the interaction between RBD and ACE2 (Fig. 2).
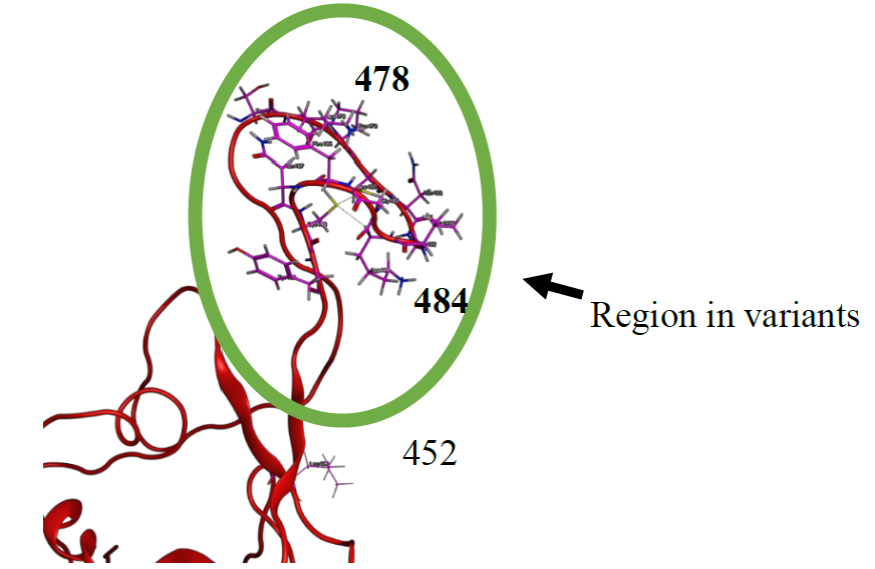
Fig. 2 RBD (Red) shows amino acids Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488 and, Tyr489 (Pink), and the positions of 478 and 484 in SARS-CoV-2 variants.
The docking is focused on the SARS-CoV-2 variants of concern from the end of 2021, such as the Delta (T478K and E484Q) and the Omicron (T478K and E484A), but other SARS-CoV-2 variants have presented the mutation in the position 484 (Beta, Gamma, Kappa, Eta) [50]. So, this region between 477 to 489 in RBD is important to interact with ACE2. Recently (January 2022), a new wave has emerged due to the Omicron variant, where the characteristics of the mutations in 478 and 484 show us that in the future other SARS-CoV-2 variants could be developed, and that they could use this region as a mechanism to evade the therapeutic options that have been developed.
In this study, a methodology developed in our drug research group was used, which has had advances in different developments of new drugs [38, 53-56]. Carrying out the selection of ten compounds, analyzing the ΔGbinding of 30 conformers of each compound, it gives us a greater probability of selecting the compounds that could be interacting between the amino acids: Ser477, Lys478, Pro479, Cys480, Asn481, Gly482, Val483, Lys484, Gly485, Phe486, Asn487, Cys488, and Tyr489, in the RBD (Fig. 1). Subsequently, the compounds selected evaluated them by toxicity prediction web servers (Table 2 and Table S12), and each compound have acceptable values of ADME characteristics and very low probability of toxicity; which must be fulfilled by each compound selection. Thus, these results could reduce the time that must be waited for to be used in humans.
Currently there are few FDA-approved oral antiviral drugs against SARS-CoV-2; Paxlovid (Pfizer) and Molnupiravir (Merck), it shows the necessity to continue with efforts to develop drugs against this disease. The results of this study could be complemented with other compounds targeting the region nearly to position 501 in the RBD [24-26, 28], due to it is reported that this region (Ser494, Gln498 and Asn501 in RBD) could destabilize the interaction with ACE2 [51], so that by this manner; using compounds directed to two regions in the RBD could block the interaction with ACE2 (Fig. 3). Therefore, this could increase the inhibitory effect and decrease the infectious process of SARS-CoV-2, as well as these compounds in combination with some of the compounds that are already proposed against ACE2 [39, 49], could stop the virus interaction with the cell receptor and avoid the formation of the SARS-CoV-2/ACE2 complex.
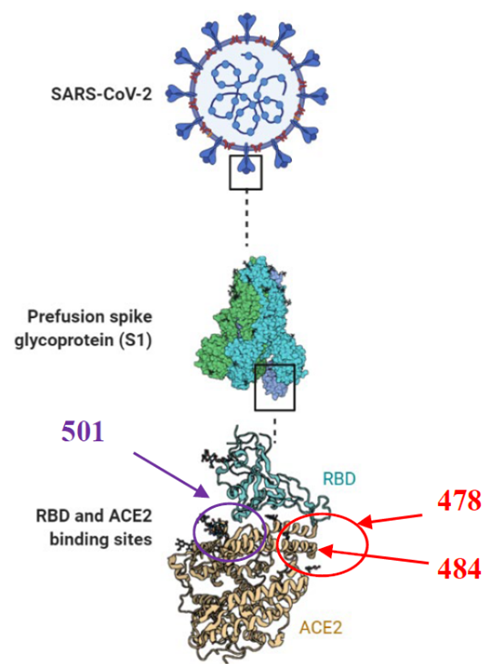
Fig. 3 Two sites for blocking interaction between RBD and ACE2. In Red the region proposes in this study nearly to 478 and 484, and in purple the region complementary nearly to 501 in the RBD. Fig created in BioRender.com.
Therefore, the results of this study could be the basis for the development of a drug that can block this region and in combination with other drugs, can achieve a better anti-viral effect. The compounds that are proposing do not have a specific use against COVID-19, nor a scientific article or registered patent, all the compounds are available to acquire them, to perform in vitro assays and to determine the effect on the interaction in the RBD of SARS-CoV-2 with ACE2.
Conclusion
This study proposes ten compounds directed to a specific region for SARS-CoV-2 variants, with a high probability to interact in the specific region in the RBD of SARS-CoV-2 (477-489), to reduce the interaction with the ACE2. As well as these ten compounds have a high probability to be safe in humans since they were validated by PreADMET server (ADME and Toxicity Predictor). These ten compounds are available at many pharmaceutical compounds synthesis company worldwide. To continue development at in vitro assays, to determine the effect of these compounds on the RBD of SARS-CoV-2 to develop a new specific adjuvant antiviral that helps combat this pandemic.

