











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Since the commercialization of the first generation of lithium-ion batteries (LIBs) in the early 1990s, this type of technology has proven to be one of the most convenient technologies for efficient energy storage and utilization. [1] For this reason, LIBs are considered the most critical milestone in the development of electrochemical energy storage technologies, and their versatility for use in portable devices, electric vehicles, and intelligent grid energy storage systems has led to increased market demand. [2] In most practical applications, high energy density, power density, efficiency in charging and discharging processes, and lifetime and safety of the devices during operation are of great importance and interest. [3] The ability of LIBs to cover such functions is determined by the rate of charging and discharging processes (determined by the reaction progress, kinetic control) and the charge storage capacity (identified as the end of the reaction, thermodynamic control). These processes are determined explicitly by the efficiency of the reversible and simultaneous reduction-oxidation processes, generating oxidation at the anode and reduction at the cathode during the discharge process. For example, the charging and discharging process of a LIB, composed of commercial LiFePO4 (LFP) and graphite (C) electrodes, where the reactions associated with these are as follows:
Oxidation reaction at the anode:
Reduction reaction in the cathode:
Global reaction:
The performance of LIBs, on the other hand, is determined by three main factors: energy density, output power density, and safety during operation. [4] The first factor, energy density, refers to the amount of energy stored per unit gram of the active materials for lithium-ion storage in the electrodes. The main strategy to achieve LIBs with high energy density has been the exploration of new active materials for advanced cathodes with high specific capacity and high operating voltages (~5 V vs. Li0/Li+), [5] some cathode materials such as LiCuxMn2-xO4 (4.9 V vs. Li0/Li+), [6] LiNi0.5Mn1.5O4 (4.7 V vs. Li0/Li+), [7] LiNixCo1-xPO4 (4.8-5.1 V vs. Li0/Li+), [8] Li2CoPO4F (5.1 V vs. Li0/Li+), [9] and Li2CuO2 (4.5 V vs. Li0/Li+), [10] which have shown good performance, keeping their structures stable during lithium ion intercalation and deintercalation processes in cycling tests.
The second factor, power density, refers to the power per volume density, which involves the rate of Li+ transference through the electrolyte during the charging and discharging processes of a cell, where secondary interfacial reactions generally take place and affect its operation, which attributed to kinetic and electrochemical stability issues, influenced by the potential drop across the interfaces, where a high proportion of the total cell resistance resides. Therefore, the cell output voltage ( ) is determined by the compatibility of all the system components, including the anode, electrolyte, and cathode, as shown in Equation 1.
where 𝛥° is the difference of standard Red-Ox potentials of the active electrode materials (𝜂𝑐)𝑐 and (𝜂𝑐)𝑎 are the overpotentials associated with the charge transfer process at the cathode and anode, respectively; (𝜂𝑐)𝑐 and (𝜂𝑐)𝑎 are the overpotentials generated by ionic concentration at the surface of the cathode and anode electrodes, respectively; 𝑖 is associated with the cell operating current density, while 𝑅𝑖 represents the total cell resistance described by Equation 2.
where 𝑅𝑅𝑎𝑎 and 𝑅𝑐 represent the resistance to current flow at the anode and cathode, respectively. 𝑅 𝑆EIa and 𝑅 𝑆EIc represent the resistance to current flow at the solid electrolyte interphase (SEI) generated by the degradation of the electrolyte through oxidation and reduction reactions at the anode and cathode surface, respectively, and 𝑅Elect represents the resistance to current flow in the electrolyte.
Therefore, electrolyte formulations with application in LIBs must be (i) thermodynamically stable or (ii) kinetically stabilized in contact with the battery components and reaction products formed during cycling,11 including the electrodes, as mentioned above. In addition to enhanced thermal and chemical stability, the following merits for which those of liquid electrolytes based on the LiPF6 salt are known: (i) the ability to form a suitable SEI solid electrolyte interface layer on electrodes, especially on carbonaceous anodes for proper operation of LIBs, (ii) the ability to passivate the anodic dissolution of the aluminum (Al) current collector, (iii) a wide electrochemical stability window, (iv) acceptable solubility and (v) high ionic conductivity in different non-aqueous solvent systems. [12-14]
On the other hand, although many of the problems initially present in electrolytes for LIBs have been overcome, some of these have persisted and should be considered for developing specific electrolytes for new batteries with advanced chemistries. In addition to presenting themselves as viable alternatives for high-performance energy storage, they open up a new range of particular problems that can be addressed from polymer science and engineering. This review addresses the fundamental aspects of ionic conducting polymers with application to LIBs and the core concepts in designing electrolytes employed in lithium batteries with advanced chemistries.
Electrochemical characteristics of electrolytes for LIBs
From the electrochemical point of view, electrolytes for lithium-ion batteries must possess two main characteristics, high ionic conductivity values, electrochemical stability, and a desirable high lithium transfer number. Generally, each of these characteristics can be determined experimentally by independent electrochemical techniques. However, the different electrolyte systems' kinetic and ionic transport phenomena directly correlate. The clearest example is the relationship between the total ionic conductivity and the lithium transference number in single-ion conducting polymer electrolytes. In addition, the effect of the formation of anionic concentration gradients during the polarization of the electrolytes and the limitations that ionic diffusion has on their electrochemical stability. Without neglecting the correlations, the following section reviews the general electrochemical characteristics of electrolytes for LIBs.
Ionic conductivity
Ionic conductivity continues to be one of the most significant challenges in developing electrolytes for LIBs, regardless of whether they are liquids, polymerics, ceramics, gels, or hybrids. In all cases, ionic conductivity is directly proportional to the concentration of charged species, according to the following equation.
where (𝑞𝑖) is the ionic charge, (𝑢𝑖) is the mobility of each charge carrier and (𝑛𝑖) is the number of free-charged species dissolved or groups mobile charged. In an electrolytic medium, the concentration of charged species depends on the dielectric constant of the liquid or polymeric solvents. This parameter correlated with their ability to dissociate salts. In general, it is desirable to have solvents with a high dielectric constant that allow dissociating high concentrations of salts to increase the concentration of free- charged species. However, the ionic bond energies of lithium salts are a consequence of their limited solubility in the non- aqueous solvents typically used in the preparation of electrolytes for LIBs, especially when these are non-polar such as glymes or polymeric matrices. While for many liquid electrolytes, it has been considered that maintaining concentrations lower than 1M of lithium salt will prevent the formation of ionic pairs (salt clusters) that affect the free mobility of charged species, precipitation of lithium salts, and decrease of ionic conductivity, as shown in Fig. 1.

On the other hand, polymeric matrices present a relatively low dielectric constant, which strongly affects their ability to dissociate lithium salts and the mobility of ions through them. Generally, ionic transport will be favored by increasing temperature. However, it should be considered that the ionic transport mechanism in liquid electrolytes, where solvated ions are transported through the electrolyte medium, does not change with temperature. In contrast, the ion transport mechanisms in amorphous and crystalline polymer electrolytes, where the ionic transport mechanism can change from ion transport promoted by chain movement (low activation energy) to ion transport by ion hopping between different active sites of the polymer matrix (high activation energy), respectively. These ion transport mechanisms in polymer electrolytes controlling by the mobility of the polymer chains, typically characterized by the glass transition temperature ( ) parameter, which high correlated with the mobility of the polymeric matrix chains, Fig. 2 shows the typical ion conduction mechanisms in polymeric matrices.

Although this review does not deal in detail with ceramic electrolytes as pure compounds, as active or inactive fillers in polymeric matrices, it is necessary to mention that the mechanisms of ionic transport through them will be by diffusion of Li+ by direct interstitial jump, interstitial knock-off, and direct jump of vacancies (active sites in the crystal lattice), whose activation energy for the ionic conduction process will be relatively low inside the grains, but not at their boundaries. Up to this point, the possible description of mechanisms of ionic transport in liquid electrolytes, polymeric and ceramic solids and the possible combinations of these as gel-type electrolytes (polymer/liquid) and hybrid electrolytes (polymer/ceramic) will give rise to combined mechanisms of ionic transport.
Electrochemical impedance spectroscopy (EIS) measurements can quickly obtain the ionic conductivity of electrolytes. Usually, the electrolyte is placed between two blocking and inert stainless-steel electrodes to maximize the resistance to charge transfer. However, intimate contact between the electrodes and the electrolyte is essential to avoid contact problems that may affect the determination of the apparent resistance (Rb) from which the ionic conductivity (σ) is calculated (taking into account the contact area (A) and the thickness (l) of the electrolyte). To perform ionic conductivity measurements at different temperatures, in addition to guaranteeing intimate contact between the electrodes and the electrolyte, they must also be obtained by using a reliable thermal stabilization protocol that allows erasing the thermal history of the heating/cooling processes.
Chemical and electrochemical stability
All electrolytes, including ceramics, are thermodynamically unstable upon contact with electrodes, especially lithium metal. However, they are often kinetically stabilized, resulting in expansive electrochemical stability windows. To suppress the decomposition of electrolyte components and side reactions at electrode-electrolyte interfaces, the electrochemical stability window of the electrolyte (𝑉𝑜) must be within the chemical potential difference generated between the anode (μ𝐴) and cathode (μ𝐶) (Fig. 3). [15] Therefore, the working potential of the electrodes in a LIB is limited by the electrochemical stability window of the electrolyte (𝐸𝑔) (Fig. 3(a)), which is determined by the energy difference from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO); otherwise. the electrolyte can be oxidized at the anode or reduced at the cathode forming a solid electrolyte interface (SEI) on the surface of the electrodes (Fig. 3(b)). [16] For these reasons, the electrochemical stability of electrolytes has become one of the limiting factors for the use of high-voltage cathode materials in the development of advanced LIBs, which must also seek to tune the advantages offered by each of the materials that make up the anodes, cathodes, and electrolytes. [13, 17-20]
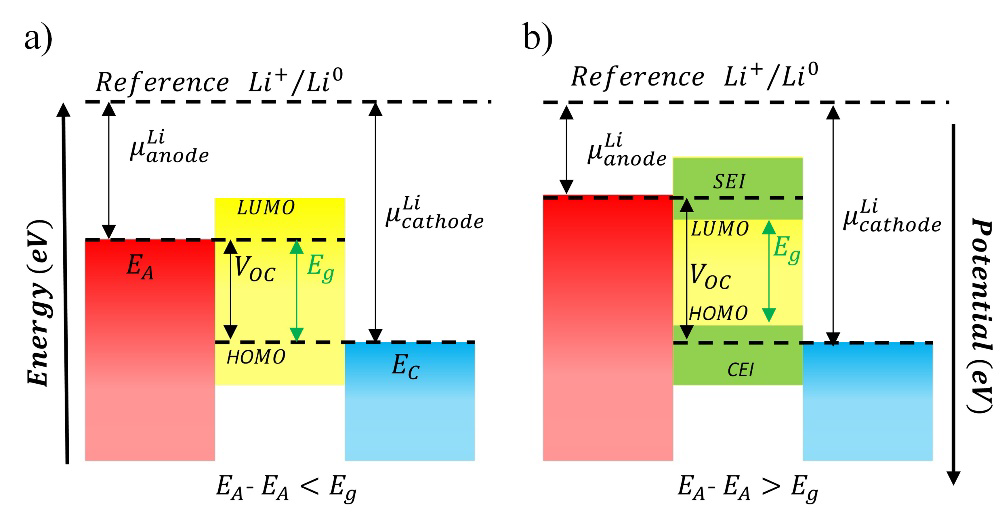
Fig. 3 The simplified three-component energy diagram. (a) Ideal electrolyte, (b) SEI formation on the electrode surface at working potentials above .
Three strategies have been implemented in liquid electrolytes to increase their electrochemical stability window. The first one is the use of additives such as lithium salts as lithium bis(oxalate)borate (LiBOB) that allows modulating and controlling the growth of the SEI layer, [21-23] as well as its reactivity with other components of the electrolytes, different types of additives such as vinyl carbonate can polymerize on the electrode surface giving rise to flexible SEI layers, [24] through which a kinetic and thermodynamic equilibrium established between the electrode-electrolyte interfaces, in addition to promoting the selective transport of ions from the electrolyte to the electrodes and vice versa. The second strategy consists of the use of cosolvents with a wider electrochemical stability window such as FEC, [11,25,26] which, despite its low boiling temperature, presents electrochemical stability up to 4.7 V vs. Li+/Li0 and improvements in ionic conductivity values, or the use of new solvents such as sulfones whose electrochemical stability values exceed 5.2 V vs. Li+/Li0, being the high melting temperature of these solvents their main limitation in practical applications. The third strategy is the use of highly concentrated electrolytes, [27,28] concentrations higher than 3 M of lithium salts in organic carbonates or lithium salts dissolved in ionic liquids, capable of maintaining the ion concentration constant during the charging/discharging periods of the LIBs, avoiding overpotentials due to ionic concentration gradients,11 which results in a decrease in the reactivity of the species that make up the electrolytes and a substantial increase in their thermal and electrochemical stability. [11]
On the other hand, both polymeric and ceramic electrolytes present intrinsic characteristics of thermal and electrochemical stability superior to liquid electrolytes. In general, all electrolytes show ion concentration gradients, especially during the charging processes of LIBs. Therefore, linear sweep voltammetry (LSV) determines electrolytes' electrochemical stability. M. Dollé et al., [29] reviewed the methods and characteristics of the measurements to be considered for evaluating electrolytes' electrochemical stability window. An asymmetric cell usually has a metallic lithium metal electrode and a blocking electrode of stainless steel or nickel. However, we could obtain a more realistic view of the electrochemical stability of electrolytes by using carbon electrodes with high surface area, [30] similar to that used in preparing standard composite electrodes for LIBs. [31] Generally, it possibly increases electrolytes' anodic and cathodic electrochemical stability. It may have different explanations, such as (i) the increase of oxidative stability of the electrolyte at higher potentials, [32,33] (ii) the shift to higher potentials of the anionic decomposition of lithium salts, [30,34] the decomposition of lithium salt anions due to their immobilization on the electrode surface by Lewis acid-base interactions, [35] (iii) ion-ion, ion-dipole and dipole-dipole interactions of the electrolyte components. [34] Mainly in the case of gel-type or hybrid polymer electrolytes with either active or inactive inorganic fillers. To alter the latter's transition energy level and raises its oxidative decomposition potential and (iv) interactions of the inorganic electrolyte with both the polymer (cross-linking sites for the EO segments) and the lithium salt anion shifting its decomposition to more anodic potentials. [36,37] However, it is necessary to consider that the electrochemical stability of the electrolytes could be modified against composite electrodes for LIBs since transition metal oxides can catalyze the oxidative decomposition of the electrolyte. [38,39]
Lithium transference number
The lithium transference number (T+) is the cation-to-anion movement ratio in a single salt electrolyte. Defined by the number of moles of lithium transferred per migration. It is often confused with the lithium transport number (t+), defined by the fraction of current carried by a specific species. This confusion by the equality of these values by considering dilute electrolytes or concentrations to ensure the absence of ionic associations or clusters of undissolved salts. [40,41] Thus, a high Li+ transference number means that the conductivity is mainly due to Li+ mobility. Since, in LIBs, Li+ is the ion involved in the redox processes at the electrodes, the concentration gradients in the electrolyte promoted by the total current intensity in the cell will be given by the migration of anions in the opposite direction to the Li+ flow, whose contribution to the polarization of the system can be reflected through a significant increase to the internal resistance of the cell. [42] For ideal electrolytes, 𝑇+=1, meaning that the anion is immobilized, and no such anion concentration gradients will form. Both ceramic electrolytes and single-conduction polymeric lithium-ion electrolytes are close to meeting this ideality condition. [43] There are several experimental techniques for determining 𝑇+. That makes no prior assumptions about the state of the analysis system; however, these techniques have other experimental or instrumental (NMR) drawbacks and are therefore relatively rarely applied. [44,45] Electrochemical methods for lithium transfer number measurements, which directly relate diffusion coefficient values to ionic conductivity values, [46] such as the use of the Nernst-Einstein equation, generate much uncertainty since the absence of ion-ion interactions must be strictly considered. However, in most practical cases, cluster formations of the form [Li2X]+ and [LiX2]- triples must be considered, in addition to the presence of [LiX] neutral ion pairs [40,47]. The method described by Evans, Vincent, and Bruce, [48] is the most common for determining Li+ transfer numbers in all types of electrolytes, but especially in polymeric and ceramic electrolytes. In this method, it is convenient from the experimental ∼point of view, in which a symmetrical Li0/electrolyte/Li0 cell is polarized by applying a potential difference (∼10 mV) until a steady state (constant current) is reached, and the transfer number is calculated by equation.
where Δ𝑉 is the applied potential difference, 𝐼0 is the initial current, 𝐼𝑠𝑠 is the steady state current and 𝑅0 and 𝑅𝑠𝑠 are the interfacial resistances before and after polarization, respectively. However, this equation is preferably valid in systems where ion-ion interactions and concentration gradients may be negligible; in concentrated systems where concentration gradients due to anionic polarization, overestimates of 𝑇+ may occur.
Classification of electrolytes for LIBs
Generally, we can classify electrolytes for LiBs into three groups for ambient to moderate operating temperatures. These are (i) liquid electrolytes (LE) (lithium salt solutions in aprotic solvents or ionic liquids), (ii) solid polymeric electrolytes (SPE) (lithium salts in polymeric matrices) and (iii) inorganic electrolytes (IE) (lithium-ion conducting ceramics) considered as single-phase ionic conductors. [49, 50] The possible combinations of these give rise to the formation of new families of two-phase ion-conducting electrolytes, classified according to their compositions and ratio into gel-type polymeric electrolytes, solid hybrid electrolytes, and quasi-solid hybrid electrolytes (Fig. 4).

Although the composition of electrolytes for LIBs has not changed significantly in recent years, a wide range of theoretical and experimental studies carried out that allow or attempt to describe phenomenologically the transport of Li+ in electrolytes, how interfaces formed at the electrode surface and how Li+ transported across them. In this section, we present the general characteristics of the different types of electrolytes, their advantages, disadvantages, and some ideas that could lead to improving the electrolytes for LIBs. Table 1 summarizes its main characteristics.
Table 1 Summary of the main characteristics of electrolytes for LIBs.
| Properties | Electrolyte types | ||||
|---|---|---|---|---|---|
| Liquid | Solid polymer | Ceramic | Gel | Hybrid polymer | |
| Ionic conductivity | good | poor | good | good | good |
| LTN | poor | Médium good | good | medium | good |
| Electrochemical stability | poor | good | good | medium | good |
| Mechanical stability | poor | good | poor | medium | medium |
| Interfacial contact | good | medium | poor | good | medium |
| Thermal stability | poor | good | good | medium | good |
Liquid electrolytes
Ionic conductivity has been the critical parameter of electrolytes, as it quantifies the amount of Li+ available for Red-Ox reactions at electrodes. The understanding of the interactions between Li+ and solvent molecules has become a relevant topic, mainly due to the discoveries about the possible variations of solvation spheres of Li+ and how these determine the chemistry of formation of electrode-electrolyte interfaces and the kinetics of Li+ migration within the formed interfaces. Until now, the preparation of liquid electrolytes consisted of dissolving lithium salts (Fig. 5) in aprotic organic solvents (Fig. 6). [14,51] Where suitable solvents should simultaneously possess high dielectric permittivity (to dissolve the salt) and low viscosity (to facilitate ion transport). Besides being inert with the rest of the cell components, they have sufficient wettability towards the electrodes, and the separator remains liquid over a wide temperature range while maintaining interfacial stability at both the anode and the cathode.
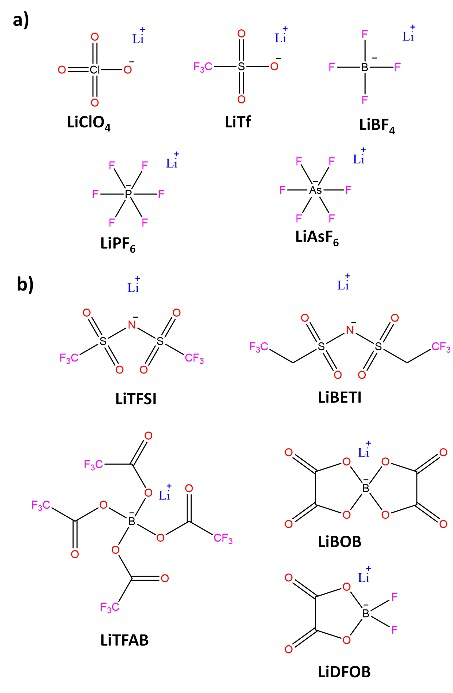
Fig. 5 Representative lithium salts for preparing electrolytes for LIBs: (a) Lithium salts known before the introduction of LIBs (b) New lithium conductive salts created for application in LIBs.
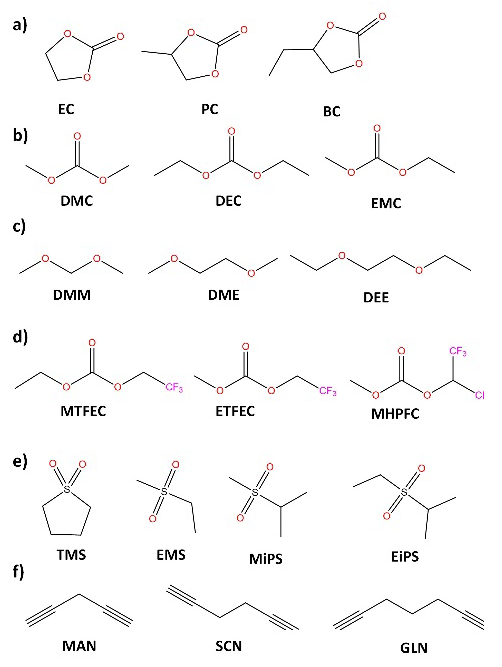
Fig. 6 Solvents for preparation of electrolytes for LIBs: (a) cyclic carbonates, (b) linear carbonates, (c) ethers, (d) partially fluorinated carbonates, (e) sulfones, (f) dinitriles.
It is desirable to find solvents with all these properties to form liquid electrolytes with binary and tertiary mixtures of solvents that confer properties. The most famous of these mixtures developed by the solvents dimethyl carbonate (DMC), diethyl carbonate (DEC), and ethyl methyl carbonate (EMC), which upon dissolving 1M of the LiPF6 salt could reach the level of 5-10 mScm-1 at room temperature, [52] giving it an operating temperature range of -30 to +60 °C. Optimization of this electrolyte could offer the feasibility to use in a wide range of battery chemistries. However, it is necessary to consider that each electrode chemistry has specific demands that can address from the point of view of varying the salt concentration, the type of anion, and its interactions with the solvent or mixture of solvents in the electrolyte. [53] Making the task of electrolyte optimization complicated from an experimental point of view, which will also have to be specific to each electrode chemistry.
Since there is a wide range of organic carbonate solvents, carboxylates, glymes, [54-56] sulfones [57], sulfoxides, [58] and nitriles, [59] can be used to prepare liquid electrolytes (Fig. 6). [14] However, the understanding of the ionic transport mechanisms and special characteristics of different types of lithium salts and solvents is relevant for the understanding and design of polymer electrolytes, since to a large extent, many of the properties of solvents and their behavior towards different lithium salts can be extrapolated to polymeric matrices containing functional groups with structural similarities.
Solid polymer electrolytes
The use of polymer electrolytes in LIBs presents essential advantages, such as safety during operation, ease of synthesis, low production cost, good compatibility with lithium salts, and excellent mechanical, chemical, and electrochemical stability. [60] Poly(ethylene oxide) (PEO) based electrolytes are considered promising candidates for use in high energy density solid-state LIBs, [61] however, classical linear PEO does not meet the ionic conductivity requirement needed for implementation σ > 10-4 S cm-1 at 60 °C, due to the formation of semi-crystalline domains that limit of movement of the polymer chains especially at low temperatures. [62,63] In this regard, different research groups have explored different approaches to increase the ionic conductivity of classical PEO-based electrolytes: (i) physical blends of polymers such as poly(vinylidene fluoride) (PVDF), [64] poly(acrylonitrile) (PAN), poly(vinyl alcohol) (PVA) and polylactic acid (PLA) [65] among others (Fig. 7). And (ii) chemical synthesis of PEO derivatives such as random and block copolymers and branched and cross-linked polymers. [61]

Polymeric matrices form classical solid polymer electrolytes (SPEs), generally based on poly(ethylene oxide) (PEO)-derived structures and dissolved lithium salts (e.g., Li+ X-: EO; X: PF6-, ClO4-, TFSI-, CF3SO3-) of relatively small anions. [43,60,66,67]The coordination sites within the polymeric structure determine Li+ transport. Therefore, Li+ moving accompanied by the temperature-dependent micro -Brownian motion of the polymeric chains, which manifested from the glass transition temperature ( ). [68] Whereas the ionic conductivity in those classical SPEs visualized as the combination of cooperative ion/polymer motion with occasional independent ion motions. [69] The anion does not significantly interact with the polymer chains, but its motion requires a free volume between polymeric chains. A natural structural consequence of polymer electrolytes and the ionic conduction mechanism is that anions are more mobile than cations. [69,70] This is reflected as the dominant contribution of anions to the charge transfer process, which can be associated with Li+ coupling to Lewis-type basicity sites in the polymer matrix. [71,72] The relatively low ionic conductivity values (between 10-5 and 10-6 S cm-1) and lithium-ion transference numbers 𝑇+, generally, less than 0.5, due to the simultaneous movement of anions and cations, [66,73] and the poor interfacial properties of SPEs, remain deterministic barriers to their application in large-scale batteries.
The increase of 𝑇+ in SPE can be achieved through the immobilization of anions in the polymeric chains, obtaining single lithium-ion conducting polymeric electrolytes capable of exhibiting lithium transfer number values 𝑇+ close to unity, making them attractive for use in LIB, [74] although their conductivities are lower than those of dual-conducting polymer electrolytes. [72,75,76] Single lithium-ion conducting polymer electrolytes (SLICPE) have been synthesized from precursors with anions capable of charge delocalization: i) based on bis(sulfonyl) imide, [77,78] ii) based on perfluoro ether sulfonate, [78,79] and iii) based on sp3 - coordinated boron atoms, [80-82] among others. [83,84] The synthesis of SLICPE based on boron atoms is among the simplest and with the lowest cost. [85,86]
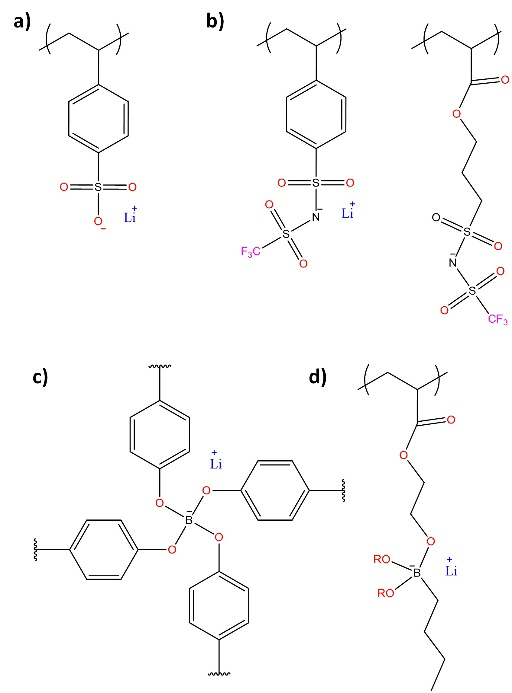
Fig. 8 Representative structures of lithium ion-bonded conducting polymeric electrolytes (SLICPE): (a) sulfonates, (b) sulfonimides, (c) tetracoordinate cross-linked borates, and (d) asymmetrically tetracoordinate borates.
On the other hand, polymeric binders have a fundamental role in the control of microstructures due to their function as an effective dispersion agent to connect the electrode species, adhering them strongly to the current collectors and between the particles. In addition, binders are the key component for the control of covering the active material particles; likewise, Li+ solvation processes are controlled by the nature of the binder and have a great effect on the stability of this interface. [36,87,88] This is especially true for composite electrodes where each of the components provides specific functionality. For proper electrochemical performance, cooperative interaction between the electrode components needs to be achieved to maximize the contributions of ionic and electronic conductivity. Several recent reports have shown those electrode capacitance limitations associated with fast charging and discharging processes arise from low ionic and electronic conductivities, i.e., restrictions. On the one hand, for Li+ diffusion through porous electrodes, and on the other hand, for electronic transport from the current collector to the active material particles, through the electronically conductive additive. [89- 91]
Ceramic (inorganic) electrolytes
Some reviews focused on the study of inorganic electrolytes that address preparation processes, structural characteristics, and a detailed understanding of the intrinsic factors that determine their electrochemical properties. This review gives a brief presentation of these materials to give a broader perspective of the different types of electrolytes that can use to develop advanced LIBs. Inorganic electrolytes are classified into soft sulfides and rigid oxides or grouped according to their structural characteristics into crystalline, glassy, and glass-ceramic. These generally have excellent in -grain ionic conductivity values. However, their high resistance to ionic transport at grain boundaries limits their practical usefulness as pure components in preparing electrolytes for LIBs.
On the other hand, vitreous and glass-ceramic materials have recently generated great interest mainly due to their isotropic ionic conduction without grain boundary resistance and their amorphous nature, which has encouraged their use as adequate solid- state electrolytes for LIBs. The transport of Li+ in ceramics is highly dependent on defects, classified as point, linear, planar, bulk, and electron. Generally, ion transport (ionic diffusion) consists of random walks along an energetically static path of separate moving point defects generated by thermodynamic equilibrium. The movement of Li+ is mainly by vacancy, interstitial, and interstitial-substitutional exchange mechanisms. These Li+ transport mechanisms depend on three factors: type of carrier, pathway, and type of diffusion.
In general, in lithium-ion conducting ceramics, the grain boundary resistance is much higher than the intragrain resistance. The structure and composition of the grain boundaries can be very different in each type of crystal. Conduction along grain boundaries is a limiting factor in most ceramic electrolytes, increasing the activation energy of ion transport and reducing the overall ionic conductivity. Some authors have attributed ion transport limitations at grain boundaries to Li+ depletion in the surface layer, generated by structural and chemical modification, significantly different from that present in the grain interior, which disfavor energetically favorable Li+ storage and transport conditions. Although compared to liquid electrolytes, inorganic solid electrolytes (ISE) suffer from ionic conductivity, some NASICON-like materials (e.g., Li1.3Al0.3Ti1.7(PO4)3), [92,93] perovskites (e.g., La0.57Li0.29TiO3), [94-96] garnet (e.g., Li7La3Zr2O12), [97,98] and sulfidic electrolytes (e.g., Li7P3S11) [99,100] have achieved desirable values of ionic conductivity at room temperature, as well as exhibiting chemical stability at atmospheric conditions and electrochemical stability at high oxidation voltages. However, most are unstable upon contact with lithium metal and highly fragile. Mainly, sulfidic ceramic electrolytes exhibit the highest ionic conductivities but, at the same time, sensitivity to air and moisture. [100-103] Garnet-type ceramics are stable in contact with lithium-metal but react with water to form decomposition products that deposit at their interface. [97,98] Perovskites possess high mechanical strength and resistance to high oxidation potentials but are unstable in contact with lithium metal and have poor ionic conductivity at grain boundaries. [92,96]
After this brief review of the characteristics of the different single-phase Li+ conducting electrolyte systems defined. It is possible to state that single-phase electrolyte materials alone lack one or some of the desirable attributes in electrolytes, judged by the limited electrochemical stability of liquid electrolytes, poor conductivity of polymer electrolytes, and poor chemical and mechanical stability of ceramic electrolytes. They are limited in electrochemical performance and processability. The Li+ conducting two-phase electrolytes, which are constituted by the combinations of polymer-liquid, polymer-ceramic, and ceramic-liquid electrolytes, give rise to the so-called gel- type polymer, hybrid solid polymer, and hybrid quasi-solid electrolytes, [104] respectively. The wide range of electrolyte materials that can be selected for the preparation of such ion-conducting two-phase electrolytes opens the prospect for their possible application in different types of battery systems such as the so- called lithium-ion batteries of advanced chemistries like lithium-metal, lithium-sulfur, and lithium-oxygen. A brief review of the main characteristics of these lithium-ion conductive two-phase electrolytes is presented below.
Gel polymer electrolytes
Gel-type polymer electrolytes (GPE) have the advantage of combining the good ionic conductivity and wettability properties of liquid electrolytes (although there are safety issues such as leakage, flammability, and electrochemical stability) with the good mechanical properties of solid polymer electrolytes. Thus, GPEs have attracted great interest as electrolytes in high-performance LIBs. However, the interfacial properties between electrodes and electrolytes must be taken into account when replacing the liquid electrolyte with GPE. Therefore, developing an electrolyte that simultaneously has high ionic conductivity, interfacial properties, good mechanical properties, and thermal and electrochemical stability remains a challenge.[105,106]
In general, GPEs are composed of polymeric matrices, liquid solvents such as plasticizers, lithium salts, and additives such as inorganic fillers. Where, commonly the polymeric matrices that provide mechanical strength are composed of the polymers PEO, PAN, PVDF, poly(vinylidene-co-hexafluoropropylene fluoride) (PVDF-HFP), and poly(methyl methacrylate) (PMMA). Plasticizers are solvents such as carbonates (propylene carbonate (PC), ethylene carbonate (EC), dimethyl carbonate (DMC), and diethyl carbonate (DEC), ethers (tetraethylene glycol dimethyl ether (TEGDME), 1,2-dioxolane (DOL), dimethoxymethane (DME) and ionic liquids. Dissolved lithium salts and mixed lithium single-conducting polymers are the sources of Li+ preferentially transported by liquid plasticizers. One of the advantages of GPEs over electrochemically inert SPE is that they can react with the electrode surface thanks to plasticizers that form stable, easily modulated SEI layers similar to those obtained when using liquid electrolytes. On the other hand, plasticizers decrease the resistance to ionic transport and result in poor thermal stability and associated safety risks, such as fires and explosions during thermal runaways. Therefore, it is necessary to perform a GPE optimization process to achieve better overall performance in each type of LIBs with advanced chemistries. Starting with the correct selection of the components, depending on the characteristics of the electrode components and the required operating conditions. Followed by the optimization of the ratio of the components according to their electrochemical performance without neglecting the mechanical and thermal properties of the GPEs. [106]
There are different methods of preparation for GPE. [107] The most popular consists of dissolving a polymeric matrix in organic solvents of low boiling point, then plasticizers and lithium salts added, and failing that, inorganic fillers, and then the solvent evaporated. In this type of GPE, it is vital to swell the polymeric matrices and control the level of drying of the GPE. The GPE prepared by physical mixtures generated due to the weak electrostatic interactions between the different components should consider. [106] Therefore, by increasing the operating temperature probably, the energy of the thermal agitation will be enough to break the electrostatic interactions, [47] and consequently, the GPE will be transformed into a liquid electrolytic mixture containing lithium salts and plasticizers.
On the other hand, in the chemical methods of GPE preparation (radiation thermal or electrochemical), [108-110] also called in situ preparation methods, the cross-linked monomers, initiators, plasticizers, and lithium salts dissolved to form a liquid electrolyte solution in a specific ratio to develop a precursor solution. [110] Subsequently, the polymerization process is initiated under specific reaction conditions to form a cross-linked polymeric matrix, in which pores or nanopores of the liquid electrolyte solutions are immobilized homogeneously to obtain GPE. [76, 86] In addition, the formation of covalent bonds in the polymeric matrix generates a strong cross-linked structure, which exhibits excellent thermal stability without solvent leakage at high temperatures or long-term aging.
Hybrid polymer electrolytes
Hybrid polymer electrolytes (HPEs) for LIBs comprise an inorganic, ionic, or non-ionic conductive component. [111] This type of electrolyte tries to take advantage of liquid and polymeric electrolytes offers, along with improving ionic conductivity and electrochemical stability properties. [112,113]
The first hybrid polymer electrolytes were prepared by adding micro and nano ceramic particles such as Al2O3, TiO2, and SiO2 (considered inactive ceramics) to SPE, [114-118] aiming to generate new conduction pathways, increase the values of ionic conductivity, Li+ transference number and decrease the activation energy of ionic conduction processes in SPE, [119] while interfacial side reactions are inhibited at the same time, thus improving compatibility at the electrolyte-electrode interface. [120] The high surface energy of the ceramic particles generates ceramic-polymer solid interactions and ceramic-polymer ratio in HPEs, which are capable of modifying the mobility of the polymer chains and, consequently the dominant mechanism of ionic conduction. Fig. 9 shows different types of HPE classified according to their polymer/ceramic ratio, the blue lines refer to the favored routes for the ionic conduction process, where the evident existence of different mechanisms and activation energies requires a process of optimization of the polymer/ceramic ratios, called percolation analysis of ionic transport. [121]
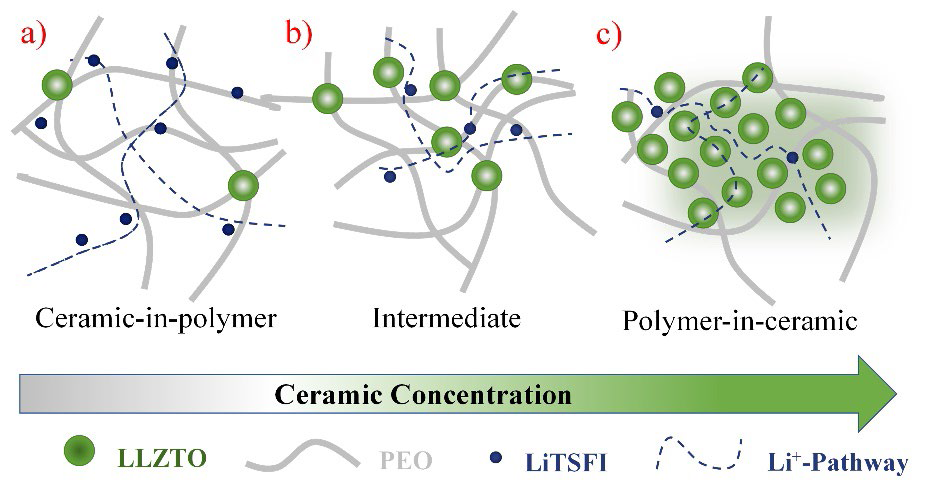
Fig. 9 Schema for (HPEs): ceramic/polymer solid electrolyte: (a) ceramic-in-polymer; (b) intermediate; (c) polymer-in-ceramic.
The success of this type of formulation and the emergence of ion transport active inorganic materials (perovskite-type Li3.3La0.56TiO3, NASICON-type LiTi2(PO4)3, LISICON- type Li14Zn (GeO4)4, garnet-type Li7La3Zr2O12 and LIPON), was a direct step towards the formulation of hybrid electrolytes, [122] whose inorganic components possess ionic conductivity characteristics. In this way, high values of total ionic conductivity can be achieved, which is composed of the ionic conductivity of the inorganic and organic components, [123,124] either liquid or polymeric electrolytes (Fig. 9).
One of the main challenges in this type of electrolyte is to find the ionic percolation threshold between two conductive phases and to generate a homogeneous distribution of the components. It is well known that during the preparation process, phase separation is one of the main problems, either due to incompatibility, mixing problems, or precipitation of the components of higher density. [104,125] Another problem present in this type of electrolytes is associated with the compatibility of the components and preparation methods, [126,129] since the high reactivity of the active inorganic materials for the ionic conduction process can lead to a high degree of decomposition of the components and the formation of highly thick or passive interfacial layers that turn off the ionic conduction pathways, or the complete electrolyte deactivation.
Conclusions
This review describes from the electrochemical point of view the factors that determine the most relevant parameters of single-phase liquid, polymeric, and ceramic electrolytes and two-phase ion-conducting gel-type polymeric electrolytes and hybrid electrolytes, paying special attention to the factors that determine the characteristics and magnitudes of the ionic conduction processes without neglecting the importance of establishing good compatibility, kinetics, and thermodynamics between the different components of the phases and interfaces of the electrolytes and the electrodes to increase the stability and operability of the LIBs. In addition, some electrolyte materials are presented, which serve as a source for generating new electrolytes that can and should be desid according to the specific demands of each battery system with advanced chemistries.

