











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkANTECEDENTES
Los tumores cardiacos son raros durante la vida intrauterina y la infancia. La tasa aproximada de incidencia es de 0.14%.1 La prevalencia, a partir de necropsias de todas las edades, se estima en 0.0017 a 0.28%.2 Casi todos son tumores primarios y, de estos, entre 75 y más de 90% son benignos.1,3 En la mayoría de los casos las tumoraciones cardiacas son padecimientos aislados, excepto por la asociación entre rabdomiomas cardiacos y esclerosis tuberosa,1,4 sobre todo ante tumores cardiacos múltiples.5
Los rabdomiomas son el tipo histológico más común: constituyen más de 60% de los tumores cardiacos diagnosticados durante la vida intrauterina y la edad postnatal.1,6 En la vida fetal le siguen en frecuencia: teratomas, fibromas y hemangiomas.2
En los rabdomiomas se ha descrito un patrón de crecimiento bifásico, con una etapa de crecimiento habitualmente durante el final del segundo trimestre y hasta la semana 32 de la gestación.7,8 Su crecimiento durante la vida fetal se ha atribuido a la estimulación hormonal intraútero.6,8 En los años posteriores al nacimiento, algunos rabdomiomas tienen tendencia a la regresión total o parcial.7,9-11 Por ello, ciertos estudios consideran al rabdomioma cardiaco fetal no como un verdadero tumor, sino un hamartoma.12
Durante la vida fetal y neonatal temprana, los rabdomiomas no suelen originar complicaciones cardiacas,5 si bien pueden causar trastornos del ritmo cardiaco (sobre todo taquicardia supraventricular), obstrucción intracardiaca de los conductos de salida o de entrada o del foramen oval, enfermedad coronaria por compresión extrínseca o, en el caso de grandes masas, ocupación completa de alguna cavidad cardiaca.4,7,8,13-18 Por ello, pueden provocar, en casos extremos, situaciones de fracaso cardiaco, hidrops fetal e incluso muerte fetal o neonatal.4,7,9,19,20
El examen ecocardiográfico, por ser una técnica no invasiva, carente de radiación, repetible y con alta tasa de detección, constituye la mejor opción para el diagnóstico y localización de lesiones ocupantes de espacio en el corazón fetal, y ha demostrado ser útil en la toma de decisiones clínicas e indicaciones de tratamiento.21-23
En la exploración por ultrasonido, el rabdomioma aparece como una lesión nodular ovalada y de contornos bien definidos, con ecogenicidad alta y homogénea. Las tumoraciones se originan, habitualmente, en el tabique interventricular o en la pared libre auricular y ventricular, y suelen ser sésiles y con frecuencia múltiples.4,6,7,21,24 Estas características son suficientemente expresivas como para considerar que la histología no es imprescindible para confirmar el diagnóstico.1 Se ha descrito, además, la manifestación ecográfica de hipertrofia miocárdica difusa, derivada de la existencia de pequeñas tumoraciones y del efecto obstructivo de éstas.25 Una ecocardiografía normal no excluye una esclerosis tuberosa porque los rabdomiomas se encuentran en 40-60% de esos pacientes.7,26
La asociación entre rabdomiomas y síndrome neurocutáneo de la esclerosis tuberosa sucede en 30-85% de los casos, aunque no haya otros signos e, incluso, sin antecedentes familiares diagnosticados.1,13,16,17,27 La relación es más frecuente cuando hay varias masas5 (90% en tumoraciones múltiples y 10% en masas únicas).3,4,7 En más de 50% de los casos el rabdomioma es la primera y única manifestación clínica de esclerosis tuberosa durante la vida fetal y neonatal,6,17,27,28 por esto se consideran los marcadores más tempranos de la enfermedad.1,13,17
La esclerosis tuberosa es una enfermedad neuroectodérmica multisistémica rara (1/20000 en población general y 1/5800 nacimientos)29 con herencia autosómica dominante y penetrancia y expresividad muy variables.4,16,27,30 La base genética de la enfermedad está en la mitad de los casos en el locus TSC1, localizado en el cromosoma 9q34.3 que codifica la hamartina, y en 50% restante en el locus TSC2, en el cromosoma 16p13.3 y que codifica la tuberina.13,31-33 La mayoría de los casos (60-80%) se debe a nuevas mutaciones.7
La esclerosis tuberosa tiene fenotipos muy variables e impredecibles.34 Sus manifestaciones clínicas incluyen tumores en el sistema nervioso central, heterotopias de la sustancia blanca, hamartomas subependimarios, quistes o angiomiolipomas renales, linfangiomas pulmonares, lesiones cutáneas (adenomas sebáceos). Son frecuentes las convulsiones (60-80%) y el retraso en el desarrollo (45-80%).5,25
CASO CLÍNICO
Paciente de 35 años, talla de 1.68 m y 70 kg al inicio del embarazo; único antecedente médico: colitis ulcerosa tratada con mesalazina. El embarazo previo (que fue el primero) trascurrió con normalidad y terminó con el nacimiento de un varón sano. Ni ella ni su pareja refieren antecedentes familiares de interés.
El embarazo actual trascurrió con normalidad, con ecografía del primer trimestre sin anomalías, translucencia nucal de 1.5 mm, y cribado de aneuploidías de bajo riesgo. La ecografía de rutina del segundo trimestre, efectuada a las 21 semanas de embarazo, resultó insatisfactoria por mala visualización cardiaca. La paciente no se presentó al control programado.
Con 24 semanas y 4 días de embarazo la paciente acudió a reevaluación cardiaca. En esa exploración se advirtieron dos tumoraciones cardiacas de ecogenicidad aumentada y homogénea compatibles ecográficamente con rabdomiomas: una localizada a nivel apical del ventrículo izquierdo de 10 X 7 mm, con protrusión hacia la cavidad pericárdica. La otra, de 5.5 X 3.5 mm a nivel de la porción alta del septo interventricular y que afectaba la vía de salida del ventrículo izquierdo, dando lugar a estenosis subaórtica leve (pico de velocidad sistólica [PVS] 158 cm/seg) (Figura 1). También se observó un derrame pericárdico apical de 3 mm. La exploración neurosonográfica se reportó normal, sin identificación de imágenes compatibles con tumoraciones, ni áreas quísticas ni ecogénicas. Tanto el ginecólogo especialista en ecografía obstétrica como el obstetra informaron a los padres la repercusión hemodinámica condicionada por una de las tumoraciones cardiacas, así como de la posible asociación de éstas con la esclerosis tuberosa, síndrome con una penetrancia y expresividad extraordinariamente variables. Se explicó que el pronóstico perinatal quedaría condicionado por estos dos aspectos. Los padres declinaron que se efectuaran más pruebas invasivas.
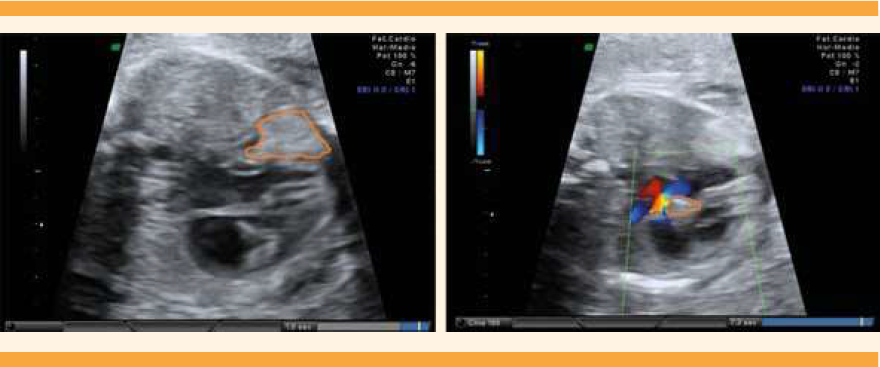
Figura 1 Ecocardiografía a las 24 semanas y 4 días de edad gestacional. Izquierda: Tumoración intracardiaca a nivel apical. Derecha: Tumoración intracardiaca a nivel de la vía de salida aórtica.
Se prosiguió con controles semanales ecocardiográficos y neurosonográficos con los que se comprobó el moderado crecimiento de las tumoraciones cardiacas y empeoramiento de la estenosis subaórtica. La neurosonografía fetal continuó normal.
A las 27 semanas y 4 días, previo consentimiento de los padres, se practicó una amniocentesis genética en búsqueda de mutaciones causantes de esclerosis tuberosa. En ese momento las tumoraciones medían 17 x 7.4 mm y 7.8 x 7.6 mm (Figura 2), con estenosis aórtica moderada (PVS 160-180 cm/seg), con 4.2 mm de diámetro valvular aórtico. Figura 3

Figura 2 Ecocardiografía a las 27 semanas y 4 días de edad gestacional. Izquierda: crecimiento de la vía de salida aórtica. tumoración intracardiaca a nivel apical. Derecha: crecimiento de la tumoración intracardiaca a nivel de la vía de salida aórtica.
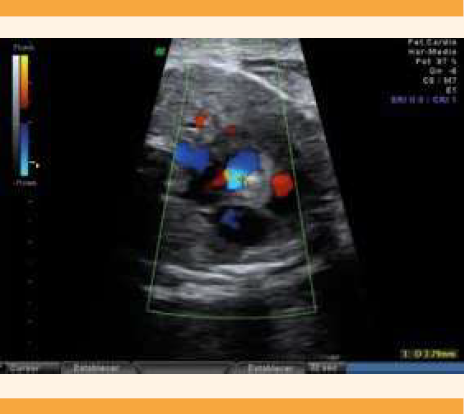
Figura 3 Ecocardiografía a las 27 semanas y 4 días de edad gestacional. Estenosis aórtica subvalvular secundaria a la tumoración intracardiaca.
El estudio genético mediante PCR cuantitativa y fluorescente (QF-PCR) reveló dos cromosomas 13, 18 y 21, y 2 cromosomas X; es decir, un feto femenino con dotación numérica normal para los cromosomas estudiados. Ese reporte se confirmó posteriormente con el análisis completo del cariotipo, que informó una fórmula cromosómica 46, XX.
El estudio genético de secuenciación confirmó la mutación en heterocigosis en el gen TSC1 (c. 1970dupC [p. Asp658ArgfsTer30]). Esta alteración no había sido previamente descrita asociada con esclerosis tuberosa en las bases de datos, ni bibliografía consultada. Sin embargo, su tipología responde a la de otras mutaciones identificadas del gen TSC1 asociadas con la enfermedad. Los análisis practicados con diferentes programas de simulación “in silico” dieron como resultado un cambio patogénico.
Ante la confirmación de esclerosis tuberosa en el feto, los padres decidieron interrumpir la gestación, por lo que, previa aprobación por el Comité de Ética de nuestro centro, se practicó el feticidio a las 28 semanas y 4 días mediante inyección intraamniótica de 1 mg de digoxina, que hizo efecto a las 48 horas. El parto se indujo con misoprostol vaginal, fue eutócico, con anestesia raquídea y nacimiento de un mortinato femenino de 1400 g.
El estudio necrópsico del feto confirmó la existencia de dos tumoraciones intracardiacas histológicamente compatibles con rabdomiomas, sin tumoraciones adicionales, malformaciones ni otros hallazgos. Se les ofreció el estudio genético de portadores en sangre periférica a los padres. Este estudio demostró que ninguno de los progenitores era portador de la variante genética encontrada en el feto.
Aspectos éticos
Se solicitó autorización a la paciente para publicar la revisión de la historia clínica y las imágenes ecográficas. Se garantizó la confidencialidad de la información para proteger los derechos de la paciente.
El diagnóstico prenatal de enfermedades graves coloca a los padres ante la posibilidad de interrumpir el embarazo. La decisión depende de muy diversos factores: gravedad de la enfermedad, posibilidades de tratamiento, semanas de embarazo, y aspectos éticos de los padres y marco legal de la sociedad en la que vive. La esclerosis tuberosa se asocia con fenotipos muy variables e impredecibles, sobre todo en cuanto al grado de repercusión neurológica, por lo que es de especial importancia el adecuado asesoramiento a los padres.
DISCUSIÓN
El diagnóstico de rabdomiomas cardiacos suele establecerse con ecografía. En los últimos años se han publicado casos de diagnóstico ecográfico en el primer trimestre de la gestación;35 el hallazgo prenatal de una tumoración cardiaca se produce, generalmente, en las exploraciones sistemáticas del segundo o tercer trimestre.4,7,9,16,36 En nuestro caso, el diagnóstico se estableció a las 24 semanas y 4 días de gestación, que pudo dilatarse por la subóptima visualización cardiaca en la semana 21 y por el retraso de la paciente en comparecer a su cita para reevaluación ecocardiográfica.
Los rabdomiomas suelen originar pocas complicaciones cardiacas durante la vida fetal y el periodo neonatal temprano.5,37 Sin embargo, en algunos casos se asocian con trastornos hemodinámicos significativos.4,7,8,13-18 Las complicaciones cardiacas obstructivas que se reflejan en este caso son, contrario a lo que suele suceder en los adultos, poco frecuentes,1,17 pero asociadas con malos desenlaces perinatales.37 En nuestro caso, el crecimiento del tumor cardiaco, situado en el tabique interventricular, condicionó el agravamiento rápido y progresivo de la estenosis subaórtica secundaria.
Debido al riesgo de complicaciones hemodinámicas y mortalidad fetal y postnatal, cuando se identifica una lesión ocupante de espacio en el corazón fetal, es necesario efectuar controles periódicos estrechos de la hemodinámica y la función cardiaca fetal.21,23 En nuestro caso, se realizaron controles ecocardiográficos y neurosonográficos semanales. Además, debe practicarse un examen sistémico en busca de lesiones en el sistema nervioso y otros órganos del feto.27 La resonancia magnética nuclear puede ser de utilidad para el rastreo del sistema nervioso central27 y determinar el grado de daño a los grandes vasos y miocardio adyacente a la masa.17 En nuestro caso, la resonancia magnética efectuada en la semana 26 de la gestación no evidenció lesiones ni otros hallazgos no detectados por ecografía.
Debido a que, en general, los rabdomiomas no suelen provocar complicaciones cardiacas, puede llevarse a cabo un tratamiento conservador de la gestación en la mayoría de los casos, con alta tasa de supervivencia (en torno al 67%).1,4,7,9,13,14,16,17 Por tanto, el pronóstico a largo plazo está principalmente condicionado por su posible asociación con esclerosis tuberosa y las impredecibles complicaciones del neurodesarrollo vinculadas con esta enfermedad.11 Si bien es frecuente que aparezcan convulsiones (60-80%) y retraso en el desarrollo (45-80%),5,25 el grado de repercusión neurológica de la enfermedad es muy variable, y hay pacientes con desarrollo neurológico normal.13,28
Las características clínicas de la esclerosis tuberosa siguen siendo el principal medio de diagnóstico de la enfermedad. A partir del Consenso de 2012 la identificación de una mutación patogénica en TSC1 o TSC2 se considera un criterio independiente y suficiente para su diagnóstico.30 Se han desarrollado técnicas de diagnóstico molecular de mutaciones de los genes TSC1 y TSC2 a partir de muestras de vellosidades coriónicas y líquido amniótico.38 Las técnicas de amplificación múltiple con sonda dependiente de ligado (MLPA) para la detección de “pérdidas” y duplicaciones en ambos genes TSC han demostrado su validez y utilidad en el estudio prenatal.38 Se consideran indicadas en historia familiar de esclerosis tuberosa y en la detección de un rabdomioma cardiaco fetal.22,39
Aunque la mayoría de los casos (60-80%) de esclerosis tuberosa aparecen por nuevas mutaciones, es importante el estudio de los familiares del paciente para descubrir formas leves de la enfermedad y proporcionar así un adecuado consejo genético.7 Se recomienda practicar el estudio de mutaciones en la unidad familiar y familiares de primer grado del feto afectado.7,22,38 En nuestro caso, se detectó en el feto una mutación en el gen TSC1, no descrita previamente. Ninguno de los progenitores resultó ser portador de la variante genética encontrada en el feto, en el estudio realizado a ambos en sangre periférica.
Si bien el reporte de este caso pretende revisar y sistematizar la conducta médica de las gestaciones con rabdomiomas, debe tenerse en cuenta que se reporta un único caso y que, debido a la realización del feticidio, no se cuenta con seguimiento posnatal.
CONCLUSIONES
Hoy día, el consejo prenatal para rabdomiomas cardiacos sigue siendo un reto porque pueden aparecer de forma aislada o asociados con esclerosis tuberosa. Las lesiones específicas cerebrales o renales asociadas con esclerosis tuberosa pueden desarrollarse progresivamente después del nacimiento. Se dispone de escasa información del desarrollo neurológico a largo plazo asociado con la esclerosis tuberosa.
El seguimiento ecocardiográfico prenatal estrecho, la planificación adecuada del parto y la gestión de complicaciones hemodinámicas pueden prevenir complicaciones neonatales.21,23,37
El diagnóstico molecular prenatal de mutaciones en los genes TSC, así como el estudio genético de la unidad familiar, deben ofrecerse como parte del consejo prenatal, ante la sospecha diagnóstica y para asesoramiento en caso de futuras gestaciones.7,22,38,39

