











 nova página do texto(beta)
nova página do texto(beta) Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkHerein, we describe an outstanding dilated cardiomyopathy (DCM) in a 16-year-old boy, whose initial diagnosis was made at 4 months of age due to failure to thrive and tachypnea. Progressive left ventricle (LV) enlargement was evident over the years. His last echocardiogram (Fig. 1, panel A, B, and C, illustrating long, short and four-chamber axis, and videos, in supplementary data) showed a LV diastolic diameter of 100 mm (z-score value of +7.9), to the author’s knowledge one of the largest ever reported. The diagnosis was also evidenced by cardiac magnetic resonance (CMR) which showed no late gadolinium enhancement (Fig. 1, panel D and E depicting coronal and sagittal views). Genetic testing identified a possible pathogenic variant in the TTN (titin) gene (c.53117 C > T). One sibling and his mother were the only first degree relatives with the same mutation, both had a normal heart and were considered asymptomatic carriers, according with a variable expressivity phenomenon. Although LV ejection fraction has always been depressed (values between 15 and 35%), his NYHA functional Class has always remained stable in I-II class. Implantable Cardioverter Defibrillator (ICD) was inserted at age 12 because of non-sustained ventricular tachycardia detected in a 24-h-Holter-monitoring. He was initiated on oral treatment with beta-blocker, furosemide, enalapril, and acenocoumarol. Two years ago, the patient was evaluated by our cardiac transplant team, but he was not listed due to his clinical stability and good functional class.
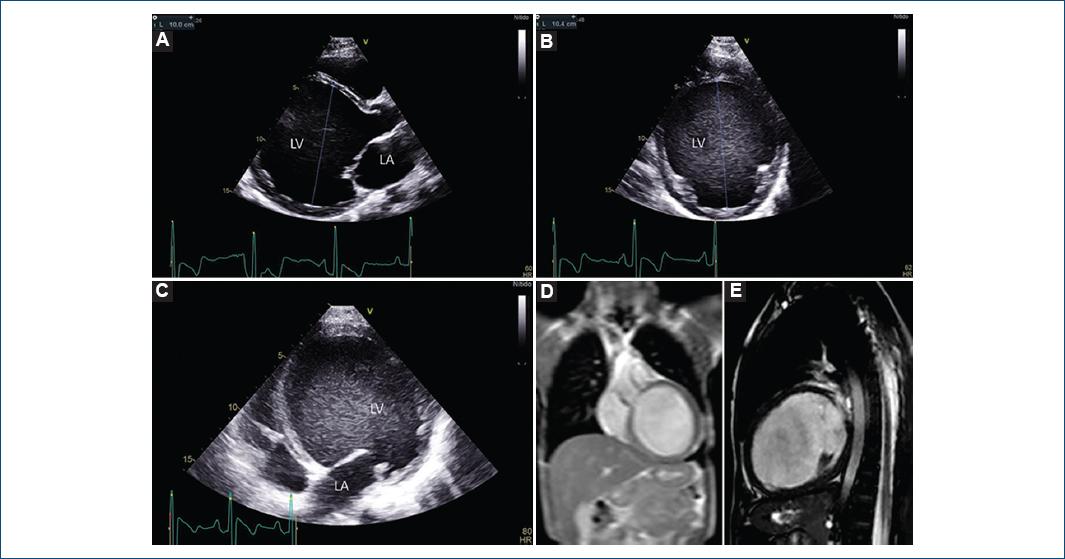
Figure 1 Echocardiography. A: long parasternal axis showing an LV end diastolic diameter of 100 mm (z-score value of +7.9). B: short parasternal axis. C: four chamber view. LA: left atrium. LV: left ventricle. Magnetic resonance imaging: D: coronal plane, E: sagittal plane.
DCM is characterized by a dilatation and impaired systolic function of the left or both ventricles1-4. It represents the most frequent cause of pediatric heart transplant4. The etiology of DCM in pediatric patients is diverse, and thus achieving the etiological diagnosis may be challenging. The known leading causes are myocarditis, inherited genetic mutations, chemical agents or toxins, nutritional deficiencies, neuromuscular diseases, autoimmune disorders, or a combination of some of these elements3,4. However, a majority of DCM cases remain with no definitive etiology. DCM could be considered as the final common response of the myocardium to diverse genetic and environmental insults1. The genetic basis of DCM is highly diverse and currently, pathogenic mutations have been uncovered in more than 60 genes1,2,5. Although several studies have pointed alterations in TTN gene as the main responsible for familial DCM, like the case here described, many other genes such as LMNA, MYH7, or TNNT2 encoding for lamin A/C, myosin, and cardiac troponin T, respectively, have also been documented3,5.
Clinical spectrum of this entity is broad, ranging from asymptomatic patients to sudden cardiac death or congestive heart failure. In spite of the astonishing heart dilation of the patient here described, the reason because he remains in functional Class I-II is probably related to a slow progression of the disease, allowing enough time to establish compensatory mechanisms for chronic heart failure. Although echocardiography represents the first innocuous, accessible and reliable diagnostic tool, CMR remains as the gold standard for a more accurate and reproducible assessment of ventricle volumes and ejection fraction as well as for evidencing myocardial fibrosis1.
The most commonly used drugs are angiotensin-converting enzyme inhibitors reducing ventricular preload and pulmonary interstitial edema, beta-blockers, and antiplatelet therapy. At the same time, mechanical circulatory support may be considered when acute refractory heart failure occurs, sometimes as a bridge to transplant3,6.
When dealing with DCM, an integrated approach with echocardiography, CMR and genetics with the corresponding medical specialists is advisable. Finding the etiological cause, to establish a precise treatment and prognosis, as well as to prevent sudden cardiac death are among the main warhorse issues. In the case described the echocardiography was the initial diagnosis tool. Twelve years later, an ICD was implanted after documenting a non-sustained ventricular tachycardia in a Holter-monitor.
Although DCM is the most common cause of heart transplant in children, in the context of a chronic severe dilated LV with depressed systolic function, conservative management may be a reasonable option if the patient remains clinically stable. Establishing the optimal timing to be listed is difficult since undergoing a heart transplant also involves drawbacks such as immunosuppressive therapy for life, risk of rejection, or a still limited long-term survival. With an unbelievable end diastolic diameter of 100 mm, we wonder whether we really know the limits of the heart. This case is an example of image and clinical discrepancy, drawing the difficult decision making that sometimes may occur in medicine.
Supplementary data
Supplementary data are available at Archivos de Cardiología de México online (10.24875/ACM.20000562). These data are provided by the corresponding author and published online for the benefit of the reader. The contents of supplementary data are the sole responsibility of the authors.

