










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Facultad de Medicina (México)
versión On-line ISSN 2448-4865versión impresa ISSN 0026-1742
Rev. Fac. Med. (Méx.) vol.56 no.2 Ciudad de México mar./abr. 2013
Monografía
Esplenomegalia
Splenomegaly
Pablo Vargas Viverosa, Rafael Hurtado Monroya, José Ángel Villalobos Alvab
a Hemato-Oncología y Medicina Interna. Hospital Ángeles del Pedregal. México, DF.
b Médico Radiólogo. Unidad de Patología Clínica. Guadalajara, Jalisco, México.
Correo electrónico: dr.pablovargas.1@gmail.com
Recibido: 28-septiembre-2012.
Aceptado: 14-enero-2013.
La esplenomegalia se define como el incremento del tamaño de bazo mayor a sus dimensiones normales, (en el adulto son 12 × 7 × 3.5 cm) con un peso aproximado de 150 g y un volumen de 300 ml (figura 1). El bazo es el órgano linfático más grande del organismo y además de participar en la respuesta inmune primaria contra microorganismos y proteínas extrañas, tiene otras funciones, entre las que destaca ser un filtro de la sangre para retirar de la circulación a los gerocitos (eritrocitos senescentes) así como a otras células sanguíneas unidas a anticuerpos. La sangre entra al bazo, se filtra a través de los cordones esplénicos y se expone a las células inmunológicamente activas.

La pulpa roja esplénica ocupa más de la mitad del volumen del bazo y es el sitio en donde se destruyen los gerocitos y se extraen las inclusiones celulares de los reticulocitos. La pulpa blanca contiene macrófagos y linfocitos B y T que participan en el reconocimiento de microorganismos y proteínas extrañas como parte de la respuesta inmune primaria (el bazo es el principal productor de IgM, sobre todo durante la infancia).
CAUSAS DE ESPLENOMEGALIA
Por frecuencia, las principales causas de esplenomegalia son la hipertensión portal por enfermedad hepática crónica, linfomas, leucemias y neoplasias mieloproliferativas, infecciones, congestión o inflamación y la trombosis de la vena esplénica.
Existen 7 mecanismos básicos que resultan en esplenomegalia:
1. Hiperplasia de las células del sistema retículo endotelial (SRE) o líneas linfoides. Es uno de los principales mecanismos de esplenomegalia. Esto ocurre en varias infecciones sistémicas, en enfermedades autoinmunes (síndrome de Felty, síndrome de Fisher-Evans, lupus eritematoso sistémico) y en tirotoxicosis. Causas infecciosas comunes de esplenomegalia incluyen endocarditis infecciosa, tuberculosis, mononucleosis infecciosa, tifoidea, histoplasmosis y paludismo.
2. Esplenomegalia congestiva. En la mayoría de los casos resulta de enfermedad hepática, en especial por cirrosis con hipertensión portal (síndrome de Banty). Hay que recordar que el hígado entre sus múltiples funciones, también es un reservorio de sangre, y cuando la fibrosis hepática progresa, se pierde esa función, por lo que la sangre se desvía (fuga) hacia el bazo y otros sitios, por lo que aumenta la presión en el territorio esplácnico. Sin embargo, la trombosis de la vena porta o esplénica y el cor pulmonale pueden tener el mismo efecto. La hiperplasia reactiva del las células del SRE de la pulpa roja ocurre con frecuencia en esplenomegalia congestiva, que aumenta aún más el tamaño del bazo. La esplenomegalia secundaria a este mecanismo es por lo general asintomática.
3. Anormalidades en la morfología de los eritrocitos. En especial esferocitosis hereditaria, talasemias y enfermedad de células falciformes, anemia hemolítica autoinmune y policitemia vera pueden causar atrapamiento de eritrocitos en los sinusoides de la pulpa roja, lo que produce esplenomegalia y anemia.
4. Hematopoyesis extramedular. Ocurre hematopoyesis esplénica en estados de insuficiencia de médula ósea como en el caso de la mielofibrosis con metaplasia mieloide, mieloptisis y osteopetrosis. En estos casos se encuentra anemia y en algunos otros reacción leucoeritroblástica.
5. Esplenomegalia maligna. Los linfomas (linfoma de Hodgkin y linfomas no Hodgkin) representan las enfermedades malignas primarias más frecuentes del bazo, aunque también es un foco de asentamientos de otras enfermedades malignas como la Leucemia Mieloide Crónica (LMC), leucemia linfocítica crónica (LLC), leucemia de células peludas (tricoleucemia) y algunas leucemias agudas. La esplenomegalia que se acompaña de adenomegalia generalizada es sugerente de Linfoma o LLC. Es muy raro que las neoplasias no hematológicas se asienten en el bazo (metástasis). Entre los carcinomas que lo pueden hacer están el de mama, pulmón, colorrectal, ovario y melanoma.
6. Esplenomegalia por depósito. La infiltración del bazo por material anormal ocurre en varias enfermedades, como la amiloidosis y varias "enfermedades por depósito" (Gaucher, Neimann-Pick). La esplenomegalia en estas condiciones con frecuencia se complica por hiperplasia reactiva de los macrófagos de la pulpa roja.
7. Otras lesiones no neoplásicas. Hemangiomas, quistes y hematomas son las entidades más comunes en esta categoría. Otras causas incluyen granulomas infecciosos por micobacterias y hongos (figura 2), granulomas no infecciosos (sarcoidosis) e infartos. Los infartos esplénicos con frecuencia ocurren en anemia de células falciformes como resultado del bloqueo de los sinusoides esplénicos por los eritrocitos deformes. También ocurre por embolismo cardiaco en el caso de endocarditis infecciosa o trombos murales. Otras causas de infartos esplénicos son la esplenomegalia masiva de cualquier etiología, principalmente secundaria a hematopoyesis esplénica, leucemia mieloide crónica y mielofibrosis primaria.

ESPLENOMEGALIA MASIVA
Así se considera al crecimiento del bazo cuyo polo inferior de encuentra hasta la pelvis, o cuando cruza la línea media hacia los cuadrantes abdominales derecho o inferiores. Son pocas las condiciones que cursan con este grado de esplenomegalia: leucemia mieloide crónica, mielofibrosis primaria o secundaria a policitemia vera o trombocitosis primaria, enfermedad de Gaucher, linfoma de células pequeñas, leucemia de células peludas, kalaazar (leishmaniasis visceral), Talasemia mayor), y el síndrome palúdico con esplenomegalia reactiva (esplenomegalia tropical).

Historia clínica
A pesar de lo extenso del diagnóstico diferencial de la esplenomegalia, la historia clínica (HC) cuidadosa y dirigida, así como un examen físico minucioso y algunos estudios de laboratorio (biometría hemática completa [BHC], pruebas de funcionamiento hepático [PFH]) reducen la lista de posibilidades a unas cuantas causas. En la HC se debe incluir especial atención en las siguientes características de la esplenomegalia y aspectos relacionados:
Aspectos principales
• Desarrollo agudo o crónico.
• Presencia o ausencia de dolor u otros síntomas locales.
• Ingestión o contacto con sustancias hepatotóxicas que resulten en hepatitis o hipertensión portal.
• Traumatismo abdominal que pueda resultar en hematoma esplénico.
• Enfermedad aguda: hepatitis, mononucleosis, paludismo, tifoidea.
• Diarrea (por salmonelosis, enfermedad inflamatoria del intestino).
• Dolor óseo, fiebre, diaforesis, malestar, fatiga, letargia, hemorragias (por su asociación frecuente con leucemia).
• Pérdida de peso, fiebre, sudoración nocturna (por su asociación con linfomas).
Antecedentes personales
• Enfermedad cardiaca: insuficiencia cardiaca congestiva, Cor pulmonale.
• Cirugías previas: infecciones quirúrgicas, trombosis, hipertensión portal.
• Transfusiones: hepatitis crónica por Virus de Hepatitis B o C (VHB o VHC), síndrome de inmunodeficiencia adquirida (sida).
• Traumatismo abdominal: posibilidad de formación de pseudo quiste.
• Viajes recientes: contacto con malaria, leishmaniasis, esquistosomiasis, tripanosomiasis.
• Conducta sexual: considerar presencia de hepatitis, citomegalovirus (CMV), virus de la inmunodeficiencia humana (VIH).
• Enfermedad hematológica conocida: deficiencia de glucosa-6-fosfato deshidrogenasa (G-6-PD), anemia de células falciformes, esferocitosis hereditaria.
Historia familiar
• Anemia, colecistectomía a edad temprana: cálculos biliares asociados a anemia hemolítica crónica.
• Esplenectomía: por anemia hemolítica.
• Origen mediterráneo: alta frecuencia de talasemia. Deficiencia de glucosa-6-fosfato deshidrogenasa (G-6-PD).
• Origen africano: alta frecuencia de a ulas falciformes, deficiencia de G-6-PD, piropoiquilocitosis hereditaria.
• Origen Judío (Asquenazí): alta frecuencia de enfermedad de Gaucher y enfermedad de Neimann-Pick.
• Origen nórdico: deficiencia de piruvato kinasa, esferocitosis hereditaria.
• Origen asiático: deficiencia de G-6-PD.
• Hemolisis aguda en hombres: deficiencia de G-6-PD.

Examen físico
La palpación del bazo se debe realizar con el examinador del lado derecho del paciente y palpar el cuadrante superior izquierdo del abdomen con la mano derecha. El paciente debe permanecer en decúbito dorsal. El examinador palpa el bazo sintiendo el margen ínferolateral. La palpación debe iniciar justo por arriba del pubis y desplazar la mano derecha hacia el cuadrante superior izquierdo del abdomen para detectar el borde medial o posterior del bazo en casos de crecimiento masivo. La percusión sobre la parrilla costal izquierda puede detectar esplenomegalia no evidente a la palpación (figura 3). En el caso de patología pulmonar obstructiva (hiperinsuflasión) se puede palpar aumento de tamaño tanto de hígado como de bazo (por otro lado normales) por desplazamiento de ambos órganos hacia abajo (pseudoesplenomegalia). Se puede confundir esplenomegalia con tumores localizados en el cuadrante superior derecho del abdomen.
Otros aspectos importantes del examen físico son los siguientes:
Piel
• Palidez: por anemia que puede indicar hemólisis, infiltración de la médula ósea, hiperesplenismo.
• Petequias, púrpura: por trombocitopenia que puede indicar enfermedad autoinmune asociada a leucemia linfoide crónica (LLC), hiperesplenismo.
• Ictericia: secundaria a anemia hemolítica, enfermedad hepática.
• Prurito: por colestasis; actividad tumoral (linfomas).
• Rash: Secundario a enfermedad aguda o crónica, Lupus Eritematoso Sistémico (LES), artritis reumatoide (AR), endocarditis infecciosa, histiocitosis.
• Eczema: por histiocitosis de Langerhans, inmunodeficiencia.
Cabeza, ojos, oídos, nariz y garganta
• Ictericia: anemia hemolítica, disfunción hepática.
• Fondo de ojo: manchas color rojo cereza en retina, opacidad de corneas (enfermedad por depósito de lípidos).
• Uveítis, iritis: sarcoidosis, AR.
Respiratorio y cardiovascular
• Disnea, fatiga: anemia, insuficiencia cardiaca congestiva, síndrome de vena cava superior (por trombosis o tumor mediastinal).
• Soplo reciente: endocarditis bacteriana.
Gastrointestinal
• Dolor abdominal: litiasis biliar, hepatitis, trauma, esplenomegalia aguda (trombosis venosa mesentérica).
• Wascitis, hipertensión portal.
• Tamaño y textura del hígado.
Musculoesquelético
• Dolor articular o artritis: LES, AR, enfermedades inflamatorias autoinmunes, gota (por hiperleucocitosis o síndrome de lisis tumoral).
• Dolor óseo: leucemia, enfermedad de Gaucher.
Neurológico
• Visión disminuida, osteopetrosis.
• Cefalea, desde síndrome anémico hasta infiltración infecciosa o maligna.
• Alteraciones encefálicas específicas por enfermedades por depósito, inmunodeficiencia, infecciones, encefalopatía hepática.
Evaluación
Imagen
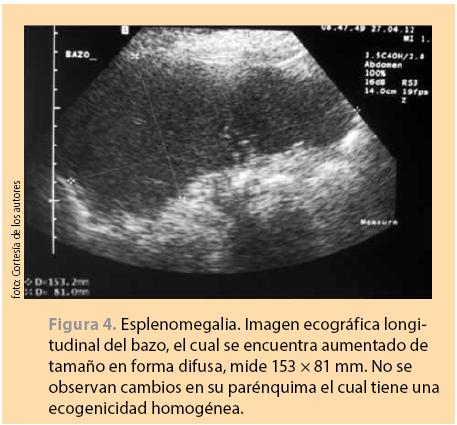
• La evaluación por ultrasonido puede confirmar la presencia de esplenomegalia o quistes, tumores, abscesos, hematomas y mide el tamaño real del bazo (figura 4).
• La tomografía axial computarizada (TAC) y la imagen por resonancia magnética (IRM) del cuadrante abdominal superior izquierdo pueden destacar con mayor fineza las anormalidades en el tamaño y la forma, y definir la patología parenquimatosa. La TAC es el estudio de elección ante la sospecha de trombosis (figura 5). La tomografía por emisión de positrones (PET-CT) con 18F-FDG, se ha convertido en una herramienta útil en el diagnóstico de lesiones de origen maligno.
• El gamagrama con Tc99m proporciona información funcional del bazo.
• La arteriografía selectiva confirma la existencia de trombosis.
Laboratorio
• La esplenomegalia en la mayoría de las veces es el resultado de una enfermedad sistémica y sólo en ocasiones orientará a un padecimiento primario del bazo, por lo que los estudios diagnósticos no se enfocan de manera directa al bazo, sino que se orientan al diagnóstico de enfermedades que cursan con esplenomegalia.
• Los estudios de laboratorio más útiles son la biometría hemática completa (BHC) con reticulocitos y cuenta diferencial, examen del frotis de sangre periférica y la prueba de función hepática (PFH).
Biometría hemática completa
• La pancitopenia puede estar presente por infiltración de la médula ósea.
• La cuenta de leucocitos puede revelar la presencia de linfocitos atípicos (irritativos) en enfermedades virales, blastos en leucemias agudas, neutropenia o neutrofilia por infección. Formas maduras mononucleares en leucemias crónicas linfoides (figura 6).
• La cifra de hemoglobina, el estudio del frotis de sangre periférica y la cuenta de reticulocitos pueden demostrar anemia, morfología anormal de eritrocitos, reticulocitosis en hemólisis o la presencia de parásitos como en el paludismo.
• La cifra de plaquetas puede indicar trombocitopenia por disminución en la producción (mieloptisis, mielofibrosis), aumento en la destrucción (autoinmunidad en LLC y linfomas, reacción a drogas, infección viral), o secuestro o hiperesplenismo.
Prueba de función hepática
• Hipoalbuminemia, tiempo de protrombina prolongado, hiperbilirrubinemia directa e indirecta (insuficiencia hepática), hiperbilirrubinemia indirecta (hemólisis), transaminasemia (daño hepático), gamma glutamil transpeptidasa y fosfatasa alcalina elevadas (colestasis).
• Anticuerpos antinucleares: LES.
• Nivel sérico de inmunoglobulinas, subclases de linfocitos T (inmunodeficiencia).
• Anticuerpos séricos para virus de Epstein-Barr (VEB), citomegalovirus (CMV), toxoplasmosis y VIH.
• Cultivos: presencia de infecciones bacterianas o micóticas; solicitar cultivos especiales.
• Estudio de trombofilia (proteína C y S, antitrombina, resistencia a proteína C activada, factor V Leiden A506G, gen de protrombina G20219A, anticuerpos antifosfolípidos y anticardiolipina): trombosis venosa mesentérica, trombosis venosa extrahepática, trombosis portal.
• Determinación de la mutación de JAK2 (V617F): pacientes con sospecha de mielofibrosis primaria o pospolicitemia vera o trombocitemia primaria (en general neoplasia mieloproliferativa cromosoma Filadelfia negativa).
• Aspirado y biopsia de médula ósea (leucemia, linfoma, neoplasias mieloproliferativas, enfermedad por depósito, infecciones diseminadas por hongos o micobacterias).
Complicaciones
Hiperesplenismo
Es un síndrome clínico caracterizado por citopenias resultantes de un estado funcional excesivo del bazo. La fisiopatología esplénica (reducción de las células sanguíneas circulantes) se atribuye a 4 posibles mecanismos: actividad fagocítica excesiva, producción de anticuerpos esplénicos que resultan en destrucción de células hematopoyéticas, hiperactividad de la función esplénica y secuestro esplénico.
Conforme el bazo crece, puede secuestrar eritrocitos, leucocitos y plaquetas y produce disminución leve a moderada en estas líneas celulares. Las citopenias severas no son frecuentes por este mecanismo y se deberá descartar otra etiología.
La obstrucción venosa es la causa más frecuente de hiperesplenismo. Cualquier incremento en la presión portal se refleja en los sinusoides venosos del bazo. Esto impide el flujo sanguíneo de los cordones y resulta en secuestro de las células sanguíneas e hiperesplenismo. La obstrucción venosa extrahepática por trombosis de vena porta es de las causas más comunes de hipertensión portal. En la obstrucción venosa extrahepática la función del hígado es normal. La obstrucción venosa intrahepática usualmente es secundaria a cirrosis.
Infarto esplénico
Los infartos esplénicos con frecuencia ocurren en anemia de células falciformes como resultado del bloqueo de los sinusoides esplénicos por los eritrocitos deformes. También ocurre por embolismo cardiaco en el caso de endocarditis infecciosa o trombos murales. Otras causas de infartos esplénicos son la esplenomegalia masiva de cualquier etiología, principalmente en mielofibrosis y LMC.
Ruptura esplénica
Se presenta en forma espontánea o con mínimo trauma en casos de esplenomegalia. Es una complicación catastrófica de aparición súbita que conduce a hemorragia masiva intra abdominal, choque y la muerte se presenta en pocos minutos a menos que se proceda de manera inmediata a la reanimación e intervención quirúrgica.
BIBLIOGRAFÍA
Aster RH. Pooling of platelets in the spleen: role in the pathogenesis of "hypersplenic" thrombocytopenia. J Clin Invest. 1966;45:645. [ Links ]
Barkun AN, Camus M, Green L, et al. The bedside assessment of splenic enlargement. Am J Med. 1991;91:319. [ Links ]
Caro J, Martínez Outschoorn U. Hypersplenism and Hyposplenism. En: Kenneth Kaushansky, Marshall A. lichman, Ernest Beutler, et al. Williams Hematology. McGraw-Hill. 8th Edition. New York 2010. Pp. 815 - 821. [ Links ]
Eichner ER, Whitfield CL. Splenomegaly: an algorithmic approach to diagnosis. JAMA. 1981;246(24):2858-61. [ Links ]
Eichner ER, Whitfield CL. Splenomegaly. An algorithmic approach to diagnosis. JAMA. 1981;246:2858-61. [ Links ]
Eichner ER. Splenic function,: Normal, too much and too little. Am J Med. 1979;66:311-20. [ Links ]
Fishman D, Isemberg DA. Splenic involvement in rheumatic diseases. Sem Arthritis Rheum. 1997;27:141. [ Links ]
Gielchinsky Y, Elstein D, Hadas-Halpern I, et al. Is there a correlation between degree of splenomegaly, symptoms and hypersplenism? A study of 218 patients with Gaucher disease. Br J Haematol. 1999;106:812. [ Links ]
Johnson JD, Raff MJ, Barnwell PA, Chun CH. Splenic abscess complicating infectious endocarditis. Arch Intern Med. 1983;143:906. [ Links ]
Porembka MR, Majella Doyle MB, Chapman WC. Disorders of the Spleen. En: John P. Greer, John Foerster, George M. Rodgers et al. Wintrobe's Clinical Hematology. Lippincott, Williams & Wilkins. 12 Edition. Philadelphia 2009. Pp.1637-54. [ Links ]
Weintraub LR. Splenectomy: who, when and why? Hosp Pract. 1994;29(6):27-34. [ Links ]
Wilkins BS. The Spleen. Br J Haematol. 2002;117:265. [ Links ]

