











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkBACKGROUND
Historically, conventional cancer treatments have primarily included surgery, chemotherapy, and radiotherapy. However, in recent years, immunotherapy has emerged as a new option1. Strengthening and reestablishing the activities of the immune system in neoplastic diseases can lead to effective responses. Specifically, various therapies activate T lymphocytes to detect and destroy cancer cells through membrane protein recognition systems called immune checkpoints2.
Programmed cell death protein-1 (PD-1) and programmed death-ligand-1 (PD-L1) are transmembrane proteins with significant functions in the regulation of the immune system. The recognition between PD-1 and its ligand, PD-L1, conveys an inhibitory signal to T lymphocytes that reduce their activation3. Under physiological conditions, the PD-L1/PD-1 interaction prevents the immune response from exacerbating. Nevertheless, in neoplastic diseases, this interaction favors the immune evasion of cancer cells4. For example, PD-L1 overexpression on lung5 and pancreas cancer cells6 allows the transmission of immunosuppressive signals. Thus, PD-1 and PD-L1 have garnered interest by the scientific community as therapeutic targets.
Several groups have reported the crystal structures of PD-17 and PD-L18. These data have determined that the interacting domains of these proteins lack cavities to host low-molecular-weight compounds. Consequently, the many attempts to develop pharmochemical inhibitors of PD-1 and PD-L1 have not been successful enough8,9. In contrast, monoclonal antibodies effectively block the immunosuppressive actions that are triggered by PD-1 and PD-L1. The inhibition of the union of PD-1, expressed in T cells, with PD-L1, expressed in tumor cells, reactivates the cytotoxic response against tumors10. Thus, in the past several years, therapeutic antibodies that target the PD-1 and PD-L1 have been developed. These drugs are now an important part of the pharmacological arsenal for many neoplastic diseases. In this review, we discuss the clinical uses of anti-PD-1 and anti-PD-L1 and their mechanism of binding to their targets, updating the status of this rapidly moving field.
ANTI-PD-1 AND ANTI-PD-L1 AS DRUGS
In the 1990s, a research group from Kyoto University, led by Honjo, discovered PD-1 while searching for genes that were involved in apoptosis. The group isolated and characterized PD-1 in murine models, showing that a genetically induced deficiency in PD-1 caused autoimmune diseases in various mouse strains. Eventually, the same function of PD-1 as a negative regulator of immune responses was shown in humans. However, the binding and functional partner of PD-1, PD-L1, was discovered until in the early 2000s11. The binding of this ligand to PD-1 on lymphocytes promotes their apoptosis and, consequently, inhibits the cytotoxic immune response. Thus, the PD-1/PD-L1 axis has an important function during the equilibrium and escape stages of tumor immunoediting12. As a result, PD-L1 is frequently overexpressed in tumor cells of many types of cancer5,6.
The finding that blockade of PD-L1 by antibodies suppresses the progressive growth of tumors in mice by restoring T-cell cytotoxicity13 implicated PD-1 and PD-L1 as targets for the development of new cancer therapies. Thus, therapeutic antibodies that target the PD-1/PD-L1 axis began to be developed immediately.
To date, there are five antibodies against PD-1 or PD-L1 that have been approved by the Food and Drug Administration (FDA) and the European Medicines Agency for use in cancer immunotherapy. Pembrolizumab (Keytruda®) and nivolumab (Opdivo®) are anti-PD-1 antibodies, both of which were approved by the FDA in 2014 for the treatment of melanoma14,15. Atezolizumab (Tecentriq®) was the first anti-PD-L1 antibody to be approved by the FDA in 2016 for the treatment of urothelial cancer and metastatic non-small cell lung cancer16. In 2017, the anti-PD-L1 antibodies avelumab (Bavencio®) and durvalumab (Imfinzi®) received approval for use against Merkel carcinoma cells and metastatic urothelial carcinoma, respectively17,18.
Eventually, all of these drugs were approved for clinical use in other types of cancer. For example, pembrolizumab is also used to treat bladder cancer, nivolumab is administered for colorectal cancer, atezolizumab is given for triple-negative breast cancer, avelumab is used for renal cell carcinoma, and durvalumab is used to treat lung cancer. The current therapeutic applications listed in Table 1 show the effectiveness of these drugs in a wide range of neoplastic diseases. Notably, in the first half of 2020, the anti-PD-1 agents, pembrolizumab and nivolumab, have received approval for a new indication.
Table 1 Clinical uses of anti-PD-1 and anti-PD-L1
| Target | Antibody | Clinical uses (year of FDA approval) | Current clinical trialsa |
|---|---|---|---|
| PD-1 | Pembrolizumab (Keytruda®) | Advanced melanoma BRAFmut (2014), BRAFneg (2015), and with the involvement of lymph nodes (2019). | Phase I: 503 |
| Non-small cell lung cancer NSCLC (2015) | Phase II: 826 | ||
| Head-and-neck squamous cell carcinoma (2016) | Phase III: 112 | ||
| Hodgkin lymphoma (2017) | |||
| Non-squamous NSCLC (2017) | |||
| Urothelial carcinoma (2017) | |||
| Unresectable or metastatic solid tumors (2017) | |||
| Gastric or gastroesophageal junction cancer (2017) | |||
| Cervical cancer (2018) | |||
| Primary mediastinal large B-cell lymphoma (2018) | |||
| Squamous NSCLC (2018) | |||
| Hepatocellular carcinoma (2018) | |||
| Merkel cell carcinoma (2018) | |||
| Renal cell carcinoma (2019) | |||
| Small cell lung cancer (2019) | |||
| Squamous cell carcinoma of the esophagus (2019) | |||
| Endometrial carcinoma (2019) | |||
| Non-muscle invasive bladder cancer (2020) | |||
| Cutaneous squamous cell carcinoma (2020) | |||
| Colorectal cancer (2020) | |||
| Nivolumab (Opdivo®) | Advanced melanoma (2014) | Phase I: 458 | |
| Squamous NSCLC (2015) | Phase II: 760 | ||
| Unresectable or metastatic melanoma BRAFmut (2015), and regardless BRAF status (2016) | Phase III: 104 | ||
| Non-squamous NSCLC (2015) | |||
| Renal cell carcinoma (2015) | |||
| Hodgkin lymphoma (2016) | |||
| Head-and-neck cancer (2016) | |||
| Urothelial carcinoma (2017) | |||
| Colorectal cancer (2017) | |||
| Hepatocellular carcinoma (2017) | |||
| Small cell lung cancer (2018) | |||
| Metastatic NSCLC (2020) | |||
| Esophageal squamous cell carcinoma (2020) | |||
| PD-L1 | Atezolizumab (Tecentriq®) | Urothelial carcinoma (2016) | Phase I: 154 |
| NSCLC (2016) | Phase II: 256 | ||
| Bladder cancer (2017) | Phase III: 70 | ||
| Triple-negative breast cancer (2019) | |||
| Unresectable hepatocellular carcinoma (2020) | |||
| Avelumab (Bavencio®) | Merkel cell carcinoma (2017) | Phase I: 87 | |
| Urothelial carcinoma (2017) | Phase II: 147 | ||
| Renal cell carcinoma (2019) | Phase III: 16 | ||
| Durvalumab (Imfinzi®) | Bladder cancer (2017) | Phase I: 165 | |
| NSCLC (2018) | Phase II: 317 | ||
| SCLC (2020) | Phase III: 42 |
aJune 30th, 2020. FDA: U.S. Food and Drug Administration.
Moreover, the five aforementioned antibodies are currently being examined in multiple clinical studies for example, clinical Stage III for pembrolizumab in multiple myeloma (NCT02579863), nivolumab in recidivist mesothelioma (NCT03063450), atezolizumab in urinary tract cancer (NCT02928406), avelumab in gastric cancer (NCT02625623), and durvalumab in cervical cancer (NCT03830866). The high number of clinical studies in progress (listed in Table 1) suggests that the clinical applications of these therapeutic antibodies will increase further19.
In addition to their blockade of the interaction between ligand and receptor to improve T-cell cytotoxicity, these drugs have other anti-tumor mechanisms. For example, avelumab increases the frequency of antigen-activated CD8+ T lymphocytes, reduces CD4+ T-cell proliferation, and induces a switch in the production from Th2 to Th1 cytokines20. Transcriptomic analyses of tumors that have been treated with nivolumab revealed that its clinical benefits correlate with increased INF-γ release by T cells and downregulation of the transporter SLC3A2, which effects a decrease in the antioxidant capacity of tumor cells, the accumulation of lipids, and the occurrence of oxidative damage and ferroptosis21,22.
Thus, it is expected that additional mechanisms that contribute to their clinical effectiveness will be reported in the coming years. In particular, future studies should analyze the effects of PD-1/PD-L1 blockade on various components of the tumor microenvironment or on specific populations of cancer cells, such as cancer stem cells. However, not all patients respond favorably to checkpoint blockade therapy and a fraction of responders relapse. It has been hypothesized that the upregulation of alternative checkpoints, such as TIM-3, and inflammatory factors in the tumor microenvironment cooperate in causing adaptive resistance by cancer cells23,24.
ADVERSE EFFECTS OF ANTI-PD-1 AND ANTI-PD-L1
Non-targeted anticancer therapies have many common adverse effects. The development of monoclonal antibodies has aimed to reduce a drugs effects on normal cells. However, the clinical use of therapeutic anti-PD-1 and anti-PD-L1 antibodies has unveiled a new set of adverse effects with various frequencies and magnitudes (Table 2). Such toxicities primarily involve the skin, endocrine glands, liver, and lung. Pneumonitis and thyroiditis appear as common side effects of anti-PD-1 therapy, whereas immune-mediated colitis and immune-mediated hepatitis are the most common adverse effects of anti-PD-L1. In general, antibodies that target PD-1 are associated with a higher severity and increased mean incidence of adverse events compared with anti-PD-L125,26.
Table 2 Most common adverse effects of anti-PD-1/PD-L1 axis antibodies*
| Antibody | Anti-PD-1 | Anti-PD-L1 | |||
|---|---|---|---|---|---|
| Pembrolizumab | Nivolumab | Atezolizumab | Avelumab | Durvalumab | |
| Adverse effect | Frequency (frequency of severe graded effects) | ||||
| Immune-mediated pneumonitis | 3.4-8.2% (0.3-3.2%) | 3.1% (not found) | 2.5% (0.8%) | 1.2% (0.5%) | 5% (1.2%) |
| Immune-mediated colitis | 1.7% (1.2%) | 2.9% (discontinued for patients with severe grade) | 20.0 % (1.4 %) | 1.5% (0.4%) | 18% (1.1%) |
| Immune-mediated hepatitis | 0.7% (0.5%) | 1.8% | 9.0% (3.0%) | 0.9% (0.7%) | 12.0% (5.0%) |
| Immune-mediated endocrinopathies | Adrenal insufficiency 0.8% (<0.4%) | Adrenal insufficiency 1.0% (not found) | Adrenal insufficiency 0.4% (<0.1%) | Adrenal insufficiency 0.5% (0.1%) | Adrenal insufficiency 0.7% (<0.1%) |
| Hypophysitis 0.6% (<0.4%) | Hypophysitis 0.6% (not found) | Hypophysitis <0.1% (not found) | Hypothyroidism 5.0% (not found) | Hypophysitis <0.1% (not found) | |
| Hypothyroidism 8.5% (0.1%) | Hyperthyroidism 2.7% (not found) | Hypothyroidism 4.6% (not found) | Hyperthyroidism 0.4% (not found) | Hypothyroidism 11% (not found) | |
| Hyperthyroidism 7% (not found) | |||||
| Hyperthyroidism 3.4% (0.1%) | Type 1 diabetes mellitus 0.9% (not found) | Hyperthyroidism 1.6% (not found) | Thyroiditis 0.2% (not found) | Thyroiditis 0.9% (<0.1%) | |
| Thyroiditis 0.6% (not found) | Type 1 diabetes mellitus <0.1% (not found) | Type 1 diabetes mellitus 0.1% (not found) | Type 1 diabetes mellitus 0.1% (not found) | ||
| Type 1 diabetes mellitus 0.2% (not found) | |||||
| Immune-mediated nephritis and renal dysfunction | 0.3% (<0.2%) | 1.2% (not found) | Not reported | 0.1% (discontinued for patients with severe grade) | 6.3% (1.4%) |
| Immune-mediated skin adverse reactions | Vitiligo 13.0% (not found) | Rash 9.0 % (not found) | Not reported | Not reported | Rash or dermatitis 26.0% (not found) |
| Vitiligo 11.0% (not found) | |||||
*Data taken from FDA prescribing information.
Several explanations have been proposed for the toxic autoimmune effects that are associated with checkpoint blockade, such as the increased activity of cytotoxic T lymphocytes against healthy tissue, the higher levels of pro-inflammatory cytokines, and greater levels of preexisting autoantibodies27. Further, some adverse effects are disease specific. For example, vitiligo, an autoimmune disorder, is a common side effect of checkpoint blockade therapy in patients with melanoma, suggesting that tumor pathophysiology is involved in the development of adverse immune effects.
BINDING MODE OF ANTIBODIES TO THEIR TARGETS
The clinical success of therapeutic antibodies that target inhibitory checkpoint proteins will engender the development of new molecules that target PD-1 and PD-L1. These molecules could be new antibodies or pharmochemicals, if the obstacles that have limited their development are overcome. These new agents must consider the available structural data of the interaction between the ligand and its receptor, as well as that of marketed antibodies with their targets. The crystal structures of PD-1 with PD-L1, in addition to each protein with their drug target, are currently available in the Protein Data Bank (PDB)28-32. These structures have revealed detailed information on the binding between proteins.
PD-1 has an N-terminal extracellular domain that is organized into 10 antiparallel beta-sheets: A, A, B, C, C, D, E, F, G, and G. Similarly, the N-terminal extracellular domain of PD-L1 has a structural arrangement of nine antiparallel beta-sheets (A, B, C, C, C, D, E, F, and G). The subdomains that mediate the binding of proteins comprise primarily the C, C, F, and G beta-sheets and their interconnecting loops, both in PD-1 and PD-L128 (Fig. 1A).
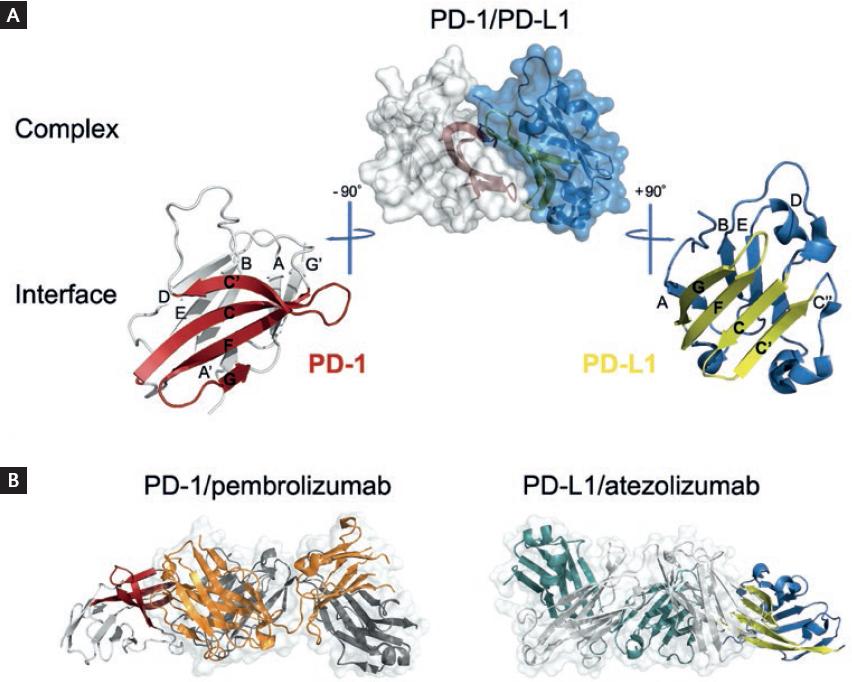
Figure 1 Relevant domains in the PD-1/PD-L1 interaction. (A) Structural representations of the extracellular N-terminal domains of PD-1 (blue) and PD-L1 (white). The names of the beta-sheets that constitute these domains are annotated. The subdomains that mediate ligand-receptor binding are highlighted in red for PD-1 and in yellow for PD-L1. (B) Structural representations of the PD-1/pembrolizumab and PD-L1/atezolizumab complexes. For simplicity, only the Fab fragments of the antibodies are shown (pembrolizumab heavy chain in orange and light chain in gray and atezolizumab heavy chain in white and light chain in green). Models were generated from crystallographic data from PDB IDs 4ZQK, 5JXE, and 5XXY.
PD-1/PD-L1 binding is effected through attractive interactions that involve 15 amino acids for PD-1 and 23-37 for PD-L1, distributed throughout the respective binding subdomains28,33,34. Because the therapeutic antibodies aim to block the interaction between proteins, their binding site lies in the same domain that mediates the ligand-receptor interaction35 (Fig. 1B).
EPITOPES RECOGNIZED BY THERAPEUTIC ANTI-PD-1 ANTIBODIES
The interactions between therapeutic anti-PD-1 antibodies and their target were revealed by the crystal structures of PD-1 in complex with the Fab fragments of pembrolizumab (PDB ID: 5B8C; 5JXE) and nivolumab (PDB ID: 5WT9)31,32. The two antibodies bind PD-1 in two regions, both overlapping partially with the binding site of PD-L1. In total, 21 amino acids in PD-1 participate in its interaction with pembrolizumab, and 14 amino acids mediate its binding to nivolumab (Fig. 2A).
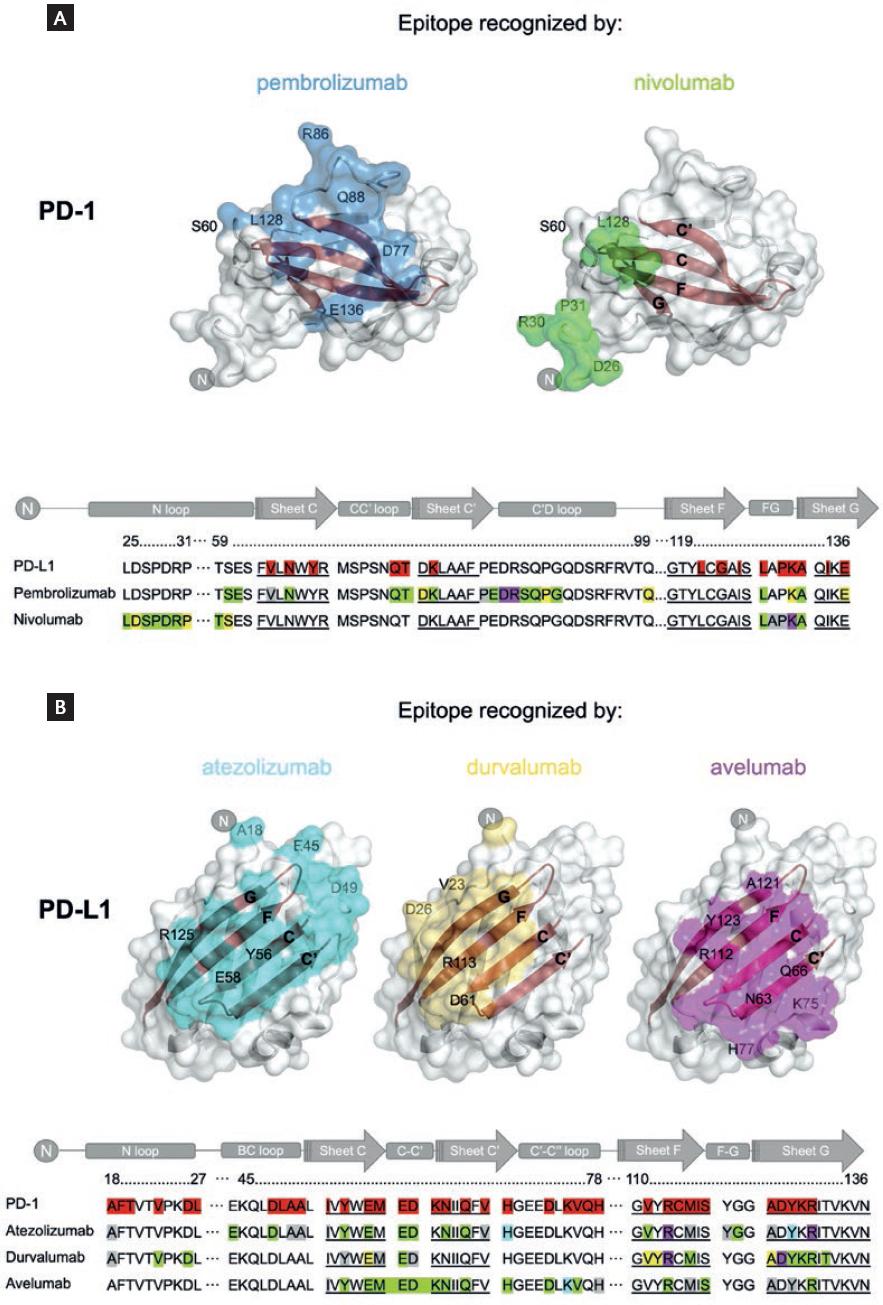
Figure 2 Epitopes recognized by anti-PD-1 and anti-PD-L1 therapeutic antibodies. (A) PD-1 structural representations (surface in white) with the epitopes recognized by pembrolizumab (blue) and nivolumab (green). (B) Structural representations of PD-L1 (surface in white) with the epitopes recognized by atezolizumab (cyan), durvalumab (yellow), and avelumab (magenta). The residues highlighted in red in the primary sequences (lower part of A and B) are those of the PD-1/PD-L1 interface. In addition, highlighted residues are those mediating the target-drug interaction: green for hydrogen bonds, yellow for water bridges, magenta for salt bridges, cyan for stacking interactions, and gray for hydrophobic contacts. For clarity, the sequence corresponding to beta-sheets is underlined. Models were generated from crystallographic data available at PDB with the following IDs: 5JXE (pembrolizumab), 5WT9 (nivolumab), 5XXY (atezolizumab), 5X8M (durvalumab), and 5GRJ (avelumab).
The epitope that is recognized by pembrolizumab overlaps with nine amino acids of the site of PD-1 that mediate the contact with PD-L1. These amino acids are distributed in the C, C, and G antiparallel beta-sheets and C-C and F-G loops. Additional interactions occur with amino acids in the C, C, G, and D antiparallel beta-sheets and the B-C, C-C, and F-G loops of PD-1. In contrast, only four amino acids in the epitope that is recognized by nivolumab overlap with the PD-1 binding site in the F-G loop. Additional interactions involve amino acids in the B-C loop of PD-1. An important contribution to the binding of pembrolizumab is made by residues in the C-D loop of PD-1, whereas for nivolumab, the amino terminal N-terminal loop of PD-1 is crucial. Notably, neither of these loops significantly participates in the binding of PD-1 to PD-L1, demonstrating that interactions beyond the receptor-ligand interfaces are important for antibody binding. In summary, the epitopes of pembrolizumab and the nivolumab differ, but both overlap partially with the PD-L1 binding site, which explains their blocking activity.
The analysis of the types of interactions that support drug-target binding indicates that 12 residues form direct hydrogen bonds and two salt bridges for pembrolizumab versus 8 and 1 for nivolumab, respectively. Thus, stronger non-covalent interactions mediate the binding of pembrolizumab. Accordingly, the binding affinity of PD-1 is (in decreasing order): pembrolizumab > nivolumab > PD-L1. The dissociation constants of PD-1/pembrolizumab and PD-1/nivolumab complexes are 27-29 pM and 1.45-3.06 nM, respectively, compared with 0.7-8.2 µM for the PD-1/PD-L1 complex31,34,36.
EPITOPES RECOGNIZED BY THERAPEUTIC ANTI-PD-L1 ANTIBODIES
The interactions between PD-L1 and anti-PD-L1 antibodies were revealed by the crystal structures that have been deposited into PDB for PD-L1 in complex with the Fab fragments of atezolizumab (PDB ID: 5XXY; 5X8L), durvalumab (PDB ID: 5X8M and 5XJ4), and avelumab (PDB ID: 5GRJ)29,33. These three antibodies bind to PD-L1 in the central beta-sheets C and F, largely overlapping with the binding site of PD-1. In total, 22 amino acids of PD-1 participate in its interaction with atezolizumab, compared with 18 with durvalumab and 20 with avelumab. The epitope that is recognized by atezolizumab overlaps by 21 amino acids with the site of PD-L1 that mediates the interaction with PD-1. Similarly, the epitope that is recognized by durvalumab overlaps by 16 amino acids. These amino acids are distributed primarily in the C, F, and, G antiparallel beta-sheets, and N-terminal and C-C loops. For atezolizumab, the overlapping region includes the beta-sheet C and B-C loop.
Notably, the epitope that is recognized by durvalumab (20 amino acids) includes the F and C beta-sheets and the C-C and C-D loops but not the N-terminal loop (Fig. 2B). For avelumab, 14 residues form hydrogen bonds versus 8 and 10 for durvalumab and atezolizumab, respectively. As expected, the binding affinity of PD-L1 is (in decreasing order): avelumab > durvalumab > atezolizumab. The dissociation constants of PD-L1/anti-PD-L1 complexes are 42.1-46.7 pM for avelumab, 22-670 pM for durvalumab, and 0.4-1.75 nM for atezolizumab30,37.
Altogether, the epitopes that are recognized by these antibodies can be useful for designing or optimizing the binding mode of new antibodies. Further, detailed knowledge of the binding mode of various antibodies will provide insights into the rational design of new smaller blockers, such as pharmochemicals and peptidomimetics.
CONCLUSIONS
Since approval of the first antibody against PD-1 in 2014, the number of clinical uses for various antibodies against inhibitory checkpoint proteins in the immunotherapy of cancer has increased14-18 and will continue to grow, considering the many ongoing clinical studies19. Their mechanism of action is blockade of the target protein, which, in turn, releases the inhibitory function of the PD-1/PD-L1 axis. Accordingly, the epitopes recognized by the antibodies partially overlap with the interfaces that mediate ligand-receptor binding. Furthermore, the epitopes of the therapeutic antibodies with a common target include shared amino acids28-32. The development of new antibodies must attack these characterized sites to maximize their probability for effectiveness. Similarly, the data on the most relevant amino acids in the formation of ligand/receptor and antibody/target complexes can be exploited to develop other types of blocking therapeutic agents, such as small molecules and peptidomimetics, which might increase the number of agents against these targets, whose relevance in multiple types of tumors has already been validated.

