











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Cushing's disease (CD) is a clinical syndrome characterized by multisystemic manifestations of hypercortisolemia, due to an adrenocorticotropin (ACTH)-secreting pituitary tumor (corticotropinoma), and is the most common cause of endogenous hypercortisolemia in adults and children1. With an estimated incidence of 0.7-2.4 cases per million per year, and a prevalence of 39.1 cases per million per year, CD is considered a rare disease2-4. Corticotropinomas represent only 15% of all pituitary tumors, but 55% of those diagnosed in 0-11-year-old patients, although only 10% of all cases occur in that age group5,6. Among adults, CD affects mostly females (3-15:1 female-to-male ratio), while males predominate among prepubertal patients2,7,8. The clinical course is usually insidious, with an estimated 3-6-year delay between disease onset and diagnosis2,7-9.
The clinical findings of adiposity with central distribution, hypertension, prediabetes or diabetes, thin skin, osteopenia or osteoporosis, hirsutism, and ecchymoses in a single patient are highly indicative of CD10. Additional manifestations are proximal myopathy, acne, purple striae, ecchymoses, dyslipidemia, hypercoagulability, amenorrhea, nephrolithiasis, dysfunction of other pituitary axes, susceptibility to infections, depression, anxiety, and, occasionally, psychosis11,12. Among adults, males might have a more severe clinical presentation and worse outcomes13. Weight gain with reduced growth velocity and normal bone age are characteristic of pediatric CD5. The diagnostic workup of CD involves an intricated algorithm, including screening tests (late-night salivary cortisol, 24 h urinary free cortisol [UFC] and dexamethasone suppression test), as well as tests for confirmation and localization (ACTH, magnetic resonance imaging [MRI], bilateral inferior petrosal sinus sampling [BIPSS], computed tomography [CT], or positron emission tomography/CT)12.
THE COMPLEXITY OF CD TREATMENT
Treatment of CD is not an easy task. Achieving prompt remission is, however, paramount, since untreated hypercortisolemia entails an estimated 5-year mortality of 50%14. Transsphenoidal surgical resection is the first line of treatment in most patients, but around one-quarter of those who achieve immediate postsurgical remission recur in the long term15,16. After reoperation, remission rates fall to 54% for persistent and 64% for recurrent CD17. Radiotherapy or radiosurgery is indicated for such cases, but their effects only become apparent after a few months or years, and their effectiveness is quite variable18. In addition, surgical reinterventions and radiotherapy increase the risk of hypopituitarism, which requires lifelong hormone replacement.
Therapeutic options for patients with persistent or recurrent disease after pituitary surgery and radiotherapy are quite limited. Bilateral adrenalectomy solves hypercortisolemia immediately and, in most cases, permanently. It entails, however, lifelong hormone replacement, as well as the risk of Nelson's syndrome19. Options for medical treatment are drugs targeting the pituitary gland, inhibitors of adrenal steroidogenesis, and glucocorticoid receptor (GR) antagonists20. The first group includes the somatostatin receptor (SST) ligand pasireotide (40% of patients controlled at 7 months with long-acting formulation), the dopamine receptor agonist cabergoline (disease control in 25-40% at 3-60 months; long-term treatment escape or intolerance in around one-quarter of patients), and temozolomide in cases with aggressive tumors or pituitary carcinoma (tumor and/or hormonal response in around two-thirds of cases)21-23.
Available adrenal steroidogenesis inhibitors are etomidate (short-term; requires in-hospital monitoring), ketoconazole, levoketoconazole, metyrapone, mitotane, and osilodrostat20,24. The glucocorticoid antagonists mifepristone (GR and progesterone receptor antagonist) and relacorilant (selective GR antagonist) are useful in the treatment of cardiovascular and metabolic effects of hypercortisolemia25,26. Retinoic acid formulations, originally considered pituitary-targeting drugs, also have effects at the adrenal level and controlled CD in around 30% of reported patients27-29. Combination schemes with pituitary-targeted and anti-steroidogenic drugs have been tested, although with variable success20. Additional potential pharmacological options are under investigation.
A NEED FOR DISEASE MARKERS
In addition to clinical improvement, normalization of late-night salivary cortisol (preferred), low-dose dexamethasone stimulation test, and/or UFC has been used to determine whether remission or disease control has been achieved in patients with CD subjected to a therapeutic intervention12. Due to the risk of late relapse, most CD patients require long-term and often lifelong periodical screening to assess the disease status15,16,30. Even more, the treatment of cardiovascular, metabolic, musculoskeletal, and psychiatric comorbidities secondary to hypercortisolemia, as well as the treatment of hormonal deficiencies, usually implies lifelong medical care31.
Chronic hypercortisolemia is detrimental and greatly impacts life expectancy. Indeed, standardized mortality ratios (SMRs) of 1.7-3.8 and of 1.6-1.9 have been estimated for patients with persistent hypercortisolemia or under remission, respectively2,3,32-34. In addition to active disease, lack of vespertine serum cortisol reduction > 40% (compared to morning cortisol) and uncontrolled diabetes mellitus predictors of mortality34. The increased mortality among CD patients is mainly due to cardiovascular complications, although infectious diseases and suicide also play a role32-36. In patients under remission, increased mortality is associated with bilateral adrenalectomy and glucocorticoid replacement33. In many cases, the cardiovascular, metabolic, and psychiatric comorbidities of CD persist even several years after remission has been achieved37. Likewise, long-term impaired health-related quality of life has been documented, which partially improves after remission38.
Unsurprisingly, patients with CD face high personal and socioeconomic costs associated with comorbidities, medical expenses, and physical and cognitive impairment39. The identification of reliable markers of disease behavior and prognosis could potentially allow for a tailored and cost-efficient management of each patient, as well as for a reduction of the medical procedure-associated stress. This would assist clinicians on deciding which patients would likely require multiple therapeutic interventions and a close follow-up, as well on identifying low-risk individuals who could be managed and screened in a more conservative manner. Over the years, various clinical, biochemical, imaging, histopathological, molecular, and genetic features have been evaluated as possible CD markers, although with variable success (Table 1 and Fig. 1).
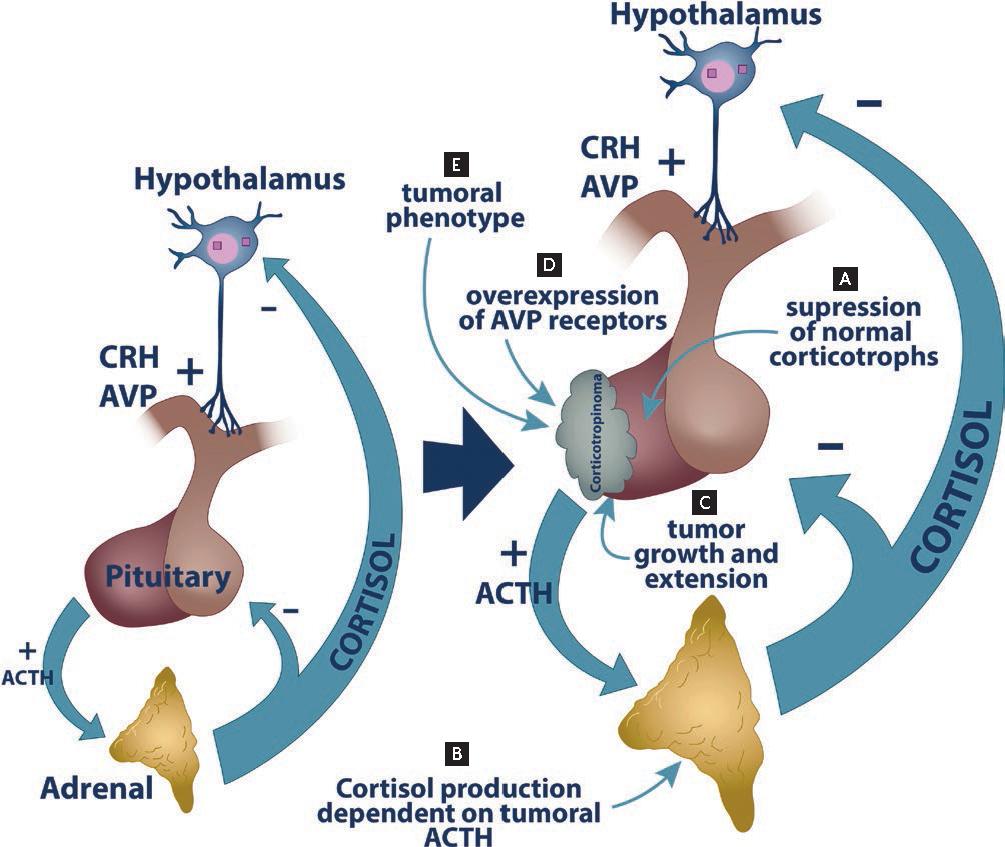
Figure 1. Multiple features of Cushing's disease (CD) are potential biomarkers. A: excessive ACTH production by a corticotropinoma suppresses ACTH secretion by normal corticotrophs. B: making adrenal cortisol production dependent on tumoral ACTH. Tumor resection causes a drastic drop in ACTH secretion that clinically presents as postsurgical hypocortisolemia, a useful marker of remission. Likewise, the time for recovery of the pituitary-adrenal axis might serve for prognosis. C: presurgical assessment of the tumor size and the degree of extrasellar extension help predict the probability of a successful resection. D: vasopressin is an ACTH secretagogue with a less prominent role than CRH under physiological conditions. In corticotropinomas, however, ACTH secretion is greatly stimulated by desmopressin, a vasopressin synthetic analogue. A measurement of this response in CD patients can be used both for diagnosis and postsurgical assessment. E: finally, the clinical and molecular tumoral phenotype in corticotropinomas is determined by a myriad of genetic defects, transcriptional alterations, and changes in protein expression. Such alterations can be determined using a combination of histopathological and molecular biology techniques, allowing for a detailed assessment of the tumor behavior.
Table 1. Summary of the reviewed potential biomarkers of Cushing's disease
| Biomarker | Meaning | Pros | Cons | Status | References |
| Biochemical parameters | |||||
| Postsurgical cortisol | Low or undetectable postsurgical serum cortisol is a well-accepted criterion of biochemical remission and low risk of recurrence | Easily available in most centers. | Variable cutoff values in literature; more robust when using cortisol ≤ 2 µg/dL | Routinely used | 16, 30, 34, 40-45 |
| Recovery of the pituitary-adrenal axis | The time required for recovery of the hypothalamus-pituitary-adrenal axis after the successful resection of a corticotropinoma is shorter in patients who experience recurrence | Evaluated and found useful in multiple studies | Requires periodical ACTH or CRH stimulation tests, not available in all centers | Requires standardization of cutoff value | 41, 48, 49 |
| Desmopressin stimulation test | The response of circulating ACTH and/or cortisol to stimulation with desmopressin might predict the probability of postsurgical recurrence | Desmopressin is available in most centers | No consensus on the dynamic test protocol | Requires standardization of protocol and cutoff values | 50-53 |
| Postsurgical ACTH | Low postsurgical plasma ACTH is an indicator of biochemical remission and probably of low risk of recurrence | Easily available in most centers | Variable cutoff values and high variability in measurements among samples | Requires standardization of cutoff values and assays | 55-60 |
| Imaging findings | |||||
| Tumor size and extension | Higher probability of postsurgical remission and lower probability of recurrence in microadenomas without extrasellar extension compared with tumors that are macroadenomas and/or have extrasellar extension | Easily available in most centers | Limited value, since most corticotropinomas are microadenomas without extrasellar extension | Routinely used | 30, 40-42, 44, 61, 62 |
| Identifiable tumor in MRI | Higher probability of successful tumor resection and postsurgical remission for patients with identifiable tumor in MRI, compared with those with no visible tumor | Easily available in most centers | Useful to predict postsurgical remission, but not long-term disease behavior | Routinely used | 40 |
| Histopathological features | Higher probability of postsurgical remission in patients without an identifiable corticotropinoma by histopathology compared with those without identifiable tumor | Easily available in most centers | Useful to predict postsurgical remission, but not long-term disease behavior | Routinely used | 44, 63, 64 |
| Granulation pattern | Densely granulated corticotropinomas occur at a younger age, are more commonly microadenomas, are less frequently invasive, and achieve immediate postsurgical remission more frequently than sparsely granulated corticotropinomas | Can be evaluated through IHC; no special equipment required | Not routinely available | Requires further investigation | 67 |
| Crooke's cell adenomas | Crooke's cell adenomas are considered a high-risk histopathological type in WHO 2017 classification | Can be evaluated through IHC; no special equipment required | Data are based on the few cases reported in the literature | Requires further investigation | 69, 71-74 |
| Pituitary carcinoma and blastoma | Pituitary carcinomas and blastomas are considered a high-risk histopathological type in WHO 2017 classification | Easily available in most centers | No markers available for early identification of carcinomas. Genetic testing for DICER1 is not widely available | Well accepted, although uncommon | 72, 75-77, 79-85 |
| Ki-67 and TP53 | Ki-67 labeling index ≥ 3% and positive TP53 immunostaining are markers of aggressive clinical behavior | Easily available in most centers | Unclear value; probably useful in combination with other markers | Routinely used; unclear clinical correlation. | (7, 4, 9-91) |
| Somatostatin receptors | SST5 immunoreactivity could be associated with functional, low grade corticotropinomas that are responsive to the treatment with pasireotide | Can be evaluated through IHC; no special equipment required | Not routinely available | Requires further investigation | 92, 98, 99 |
| Dopamine D2 receptor | DRD2 immunoreactivity could predict the response to the treatment with cabergoline | Can be evaluated through IHC; no special equipment required | Not routinely available | Requires further investigation | 100, 101 |
| CABLES1 and CDKN1B | Low CABLES1 immunostaining directly correlates with CDKN1B immunostaining; the latter inversely correlates with Ki-67 | Can be evaluated through IHC; no special equipment required | Not routinely available | Require further investigation | 103, 108-110 |
| Genetic defects | |||||
| Somatic USP8 hotspot variants | Most common genetic defect in corticotropinomas. USP8 variants seem to associate with better response to pasireotide and this genetic defect is a potential pharmacological target | Inexpensive | Requires DNA extraction and amplification and Sanger sequencing; not routinely available | Requires further investigation | 98, 99, 128, 129 |
| Gene expression | |||||
| SSTR1 and CRHR1 expression | Expression levels of SSTR1 and CRHR1 mRNA in combination with other markers could predict the probability of remission | Good predictive value in combination with other markers | Requires fresh tissue, RNA extraction and qPCR; not routinely available | Requires further investigation | 130 |
IHC: immunohistochemistry; WHO: World Health Organization.
BIOCHEMICAL PARAMETERS
Postsurgical cortisol
Because CD leads to suppression of the normal corticotroph cells, a successful corticotropinoma resection translates into transient postsurgical hypocortisolemia. Low or undetectable postsurgical serum cortisol (i.e., cortisol ≤ 1.8-5.3 µg/dL within the first 2-7 days after a surgery, depending on the series) is a well-accepted criterion of biochemical remission16,30,34,40-44. Profound hypocortisolemia (≤ 2 µg/dL) within the first 21 h after surgery accurately predicted remission in a study with a median follow-up of 11 months45. In a different study, a postsurgical cortisol ≥ 2 µg/dL translated into a 2.5-times higher risk of long-term recurrence (mean follow-up: 45 months)16. Postsurgical cortisol is one of the best validated and more easily available markers of remission and prognosis of CD. There is, however, uncertainty regarding the risk of recurrence in patients with low but not fully suppressed cortisol (i.e., 2-4.9 µg/dL)46. On the other hand, an unsuppressed postsurgical cortisol can be misleading, because around 6% of patients present delayed remission47.
Recovery of the pituitary-adrenal axis
Postsurgical hypocortisolemia is expected after a successful pituitary surgery for CD, after which most patients recover normal cortisol levels. The time required for recovery of the hypothalamus-pituitary-adrenal axis after the successful resection of a corticotropinoma is shorter in patients who experience recurrence. In one cohort, all patients with recurrence recovered the axis' function within 3 years from surgery; positive predictive values for recurrence of 64%, 61%, and 50% were calculated for patients who showed recovery within 6, 12, and 24 months, respectively41. In a second study, all patients who recurred did so within 22 months from surgery48. In the only study including only pediatric patients, there was a 14% reduction in the risk of recurrence for each month of duration of adrenal insufficiency. All patients who eventually recurred had recovered their axis within 15 months from surgery, while none of those who persisted with suppressed axis ≥ 15 months from surgery experienced recurrence49. This parameter might be a good marker of disease behavior, but it is unclear what cutoff could identify patients at the highest risk of recurrence. Furthermore, it requires periodical assessment of the axis through dynamic tests, but the required frequency is not clear, and such tests are not available in all centers.
Desmopressin stimulation test
A minimally invasive desmopressin stimulation test, measuring circulating ACTH and/or cortisol at 0, 30, 60, 120, and 180 min after the intravenous administration of 10 µg desmopressin at 8:30 AM, has been proposed for evaluating the probability of postsurgical recurrence. In one study, a threshold of ≥ 22 pg/mL or rise of 35% for ACTH and ≥ 350 nmol/L or 14% for cortisol discriminated between patients remaining in remission and those who experienced long-term recurrence50. In another cohort, an increase ≥ 27 pg/mL in circulating ACTH during the test, either during the first postsurgical week or during long-term follow-up, confidently identified patients who relapsed51. In a third study, cortisol increase of < 7.4 µg/dL was the best predictor of sustained remission52. A different study used the postsurgical disappearance of response to desmopressin as a predictor of low odds of recurrence in hypocortisolemic patients who had a presurgical response53. Unfortunately, the used cutoff values vary among studies and not all patients with CD respond to desmopressin preoperatively, which might reduce the predictive value. A combined low-dose dexamethasone suppression test and desmopressin stimulation test has been proposed as having a superior predictive value54.
Postsurgical adrenocorticotropin
Plasma ACTH suppression after surgery could also be used as a marker of remission. Cutoff values ranging from 10 to 34 pg/mL within 1-7 days after surgery have been proposed in the literature55-59. In a recent study, an ACTH nadir of < 15 pg/mL was a good indicator of both immediate postsurgical remission and remission at 3 months59. The usefulness of ACTH as a marker of long-term remission is, nevertheless, unclear60. The great variability in ACTH measurements among samples and assays, as well as the possibility of artifactual ACTH elevation due to tumor manipulation, have precluded the extended routine use of this marker in the clinic59,60.
IMAGING FINDINGS
Tumor size and extension
Most corticotropinomas are single microadenomas (tumors < 10 mm in their maximum diameter); only 5-10% are macroadenomas and the coexistence of corticotropinomas with other tumors is very rare6. According to modern series, remission after an initial surgery is achieved in 74.3-89.8% of patients with microadenomas and in 30.7-74.5% with macroadenomas in reference centers40-42,44,61,62. Recurrence has been estimated in 9.7-15.2% for microadenomas and 15.2% for macroadenomas30,40,41,61. Coincidentally, the postsurgical remission rate in patients without extrasellar extension by MRI is significantly higher than that of cases with invasion (77.1 vs. 53%)44. The use of tumor size as a predictor factor for recurrence, however, is limited, given than most corticotropinomas are microadenomas with no extrasellar extension.
Identifiable tumor in MRI
Among patients with microadenomas, those with a visible tumor by MRI harbor an 18-times higher probability of having a tumor detected during surgery, as well as a 4.1-higher probability of immediate postsurgical remission than those with no observable tumor40. Importantly, around 40% of corticotropinomas are not identifiable by MRI11. In those patients, a positive BIPSS is associated with a 93% chance of immediate postsurgical remission40.
HISTOPATHOLOGICAL FEATURES
Histopathological confirmation
Mainly due to the small size of most corticotropinomas, which might get lost during resection, and to the possibility of false-positive MRI images, around 12-19% of the patients with CD who undergo surgery do not have histopathological confirmation of a corticotropinoma44,63,64. Histopathological confirmation per se seems to be crucial for prognosis, because the postsurgical remission rate in patients without a histopathological diagnosis of corticotropinoma is only 50-69%, compared to 76-94% in patients with confirmed corticotropinomas in modern series44,63,64. Tumors that stain positive for ACTH but behave clinically non-functional are referred to as silent corticotropinomas and entail the potential for aggressive behavior6. Such tumors are not reviewed here since they do not present clinically as CD.
Granulation pattern
CAM5.2 is a low molecular weight cytokeratin antibody clone that seems to react against CK1865. Based on the granulation pattern under CAM5.2 immunostaining, corticotropinomas can be classified into two subtypes65,66. Most tumors are of the densely granulated subtype and contain abundant secretory granules that appear as a diffuse and intense ACTH staining. In contrast, sparsely granulated corticotropinomas contain scarce and small secretory granules, rendering a weak or patchy ACTH immunostaining6. Densely granulated corticotropinomas occur at a younger age, are more commonly microadenomas, are less frequently invasive, and achieve immediate postsurgical remission more frequently than sparsely granulated corticotropinomas67. Unfortunately, the role of the granulation pattern as a predictor of clinical behavior has not been extensively evaluated.
Crooke's cell adenomas
Chronic exposure to glucocorticoids leads to the accumulation, in corticotroph cells, of cytoplasmic ring-like hyaline material consisting of microfilaments that displace ACTH granules toward the perinuclear and submembranous regions68. Non-neoplastic corticotroph cells with such a characteristic appearance are known as Crooke's cells and are typically found within the normal pituitary area that surrounds a corticotropinoma69. Infrequently, corticotropinomas are themselves composed of > 50% Crooke's cells, in which case they are known as Crooke's cell adenomas70,71. These tumors might present clinically as CD or as non-functional tumors and account for 1% of all pituitary tumors and 4.4% of all corticotropinomas71.
Crooke's cell adenomas are considered a “high-risk” histopathological type of pituitary tumor in the 2017 World Health Organization (WHO) classification since they usually exhibit aggressive behavior72. Most of them are macroadenomas, usually presenting with cavernous sinus invasion and tendency for postsurgical recurrence, and even progression to carcinoma has been reported in rare cases69,73. A good response to temozolomide has been documented in a few cases; the need for this pharmacological treatment, however, has not been clearly demonstrated. Most of these tumors display strong positive immunoreactivity for TP53, but Ki-67 < 1%71. Due to their rarity, data on the behavior of Crooke's cell adenomas derive from a few case series; thus, evidence to support an increased aggressiveness for this tumor type remains inconclusive74. It is also unclear whether these tumors require a different treatment or follow-up conduct than the rest of corticotropinomas.
Pituitary carcinoma and blastoma
Other “high-risk” histopathological diagnoses are pituitary carcinomas and blastomas72. Pituitary carcinomas account for only 0.1-0.2% of all pituitary tumors75,76. These lesions, often histologically indistinguishable from pituitary adenomas, are characterized by the development of craniospinal or systemic metastases77. Most cases do not present initially with metastases, but the mean survival once these are documented is < 4 years75. Their etiology is unclear, and the possibilities of both progression of a pituitary adenoma to carcinoma and de novo arousal of carcinoma have been proposed78. Cases of ACTH-producing pituitary carcinomas, accounting for 42% of the total, display a long latency period (9.3-9.5 years) between the diagnosis of a corticotropinoma and the confirmation of metastases75. Negative immunostaining for 6-O-methylguanine-DNA methyltransferase (MGMT) might predict response to the treatment with temozolomide in patients with aggressive pituitary tumors, including carcinomas, although the predictive value of this marker is uncertain77. Although the adverse prognosis and poor response to available treatments of these lesions are well documented, there are no available markers to predict which ACTH-producing tumors will develop metastases.
Pituitary blastomas are usually diagnosed in neonates or infants, although a single childhood-onset case and one more occurring in a young adult have also been described79-81. These tumors derived from the anterior pituitary are extremely rare, poorly differentiated, and rapidly growing, almost always express ACTH and present either as clinically non-functional tumors or as CD82,83. Nineteen out of the 20 cases that have been genetically screened occurred in the setting of the autosomal dominant DICER1 syndrome, caused by germline loss-of-function variants in the DICER1 gene79-81,84,85. Even though surgical resection has been successful in some cases, 10/19 patients died during infancy or childhood due to the tumor or related complications79-81. Finding a pituitary blastoma in a patient with CD should prompt screening for germline DICER1 variants and genetic counseling85.
Ki-67 and TP53
The routine use of Ki-67 and TP53 in most centers was implemented after the 2004 WHO classification of pituitary tumors categorized tumors with a Ki-67 labeling index > 3% and extensive TP53 immunoreactivity as atypical adenomas86. Due to poor clinical correlation, however, such category was no longer included in more recent versions of the classification72,87. Nevertheless, a Ki-67 labeling index ≥ 3% is still considered a marker of aggressive clinical behavior in most centers74. Ki-67 expression is also indicated as “MIB-1 index,” since it is measured as the percentage of tumor cells that stain positive using the MIB-1 clone antibody77. This protein is implicated in the organization of heterochromatin and in the regulation of transcription, although it is not absolutely required for cell proliferation, and it is considered a measure of proliferation88.
A recent study found a high proportion of tumors with Ki-67 index < 3% among densely granulated corticotropinomas67. In a different study, higher Ki-67 values were associated with a larger tumor size89. Therefore, in corticotropinomas, Ki-67 immunostaining might be informative when used in combination with additional biomarkers. TP53 immunostaining, in combination with other biomarkers, is still considered of value in most centers, although its predictive role in CD remains unclear74,89. Loss-of-function somatic TP53 variants have recently been identified in aggressive corticotropinomas, although their long-term significance has not been determined90,91.
SST
A therapeutic option for patients with CD with an absence of postsurgical remission or recurrence is the SST ligand pasireotide. Treatment with the long-acting formulation of pasireotide normalizes UFC in 40% of patients12. The main target of this drug is SST5, although it also acts on SST1, 2, and 392. Almost all corticotropinomas express SST5 at the messenger RNA (mRNA) level, but when monoclonal antibodies are used, only 20-42% are immunoreactive for this marker93-98. It has been suggested that SST5 immunoreactivity could be associated with functional, low-grade corticotropinomas, and that this marker could predict the response to the treatment with pasireotide98,99. Its value as a predictor of clinical behavior, however, has not been formally evaluated in large cohorts92.
Dopamine D2 receptor (DRD2)
The DRD2 is expressed in the normal pituitary gland, where its main physiological function is the negative regulation of prolactin secretion through binding to hypothalamic dopamine. Since most prolactinomas retain the expression and function of DRD2, these tumors can be effectively treated with the dopamine agonist cabergoline. Around 75% of corticotropinomas also express DRD2 by immunohistochemistry, and treatment of primary corticotropinoma cultures with cabergoline in vitro leads to substantial inhibition of ACTH secretion in all tumors expressing this receptor100. Nevertheless, only around 40% of patients with CD normalize cortisol secretion after 3-6-months cabergoline treatment, and one-fifth of the patients who originally respond present escape in the long term100,101. Additional, not yet described, molecular pathways regulating DRD2 signaling might impair the response to cabergoline in these tumors.
CABLES1 and CDKN1B
A well-known feature of corticotropinomas leading to CD is their blunted response to the negative feedback exerted by the adrenal glucocorticoids102. A key player in that response is the negative cell cycle regulator CABLES1, whose expression rises in response to circulating glucocorticoids103. CABLES1 is often inactivated in human neoplasms by allelic loss, aberrant splicing, or hypermethylation104-106. Loss of CABLES1 immunoreactivity has been observed in around half of the corticotropinomas so far studied103. In addition, germline loss-of-function missense variants of CABLES1 have been reported in four patients with young-onset CD, although the heritability of the phenotype has not been demonstrated107. Interestingly, low CABLES1 immunostaining directly correlates with low immunostaining for the cyclin-dependent kinase inhibitor CDKN1B, which is frequent in corticotropinomas and has been found to inversely correlate with Ki-67103,108-110. Loss-of-function germline CDKN1B variants are associated with corticotropinomas occurring either isolated or as part of the syndrome of multiple endocrine neoplasia Type 4111. Nevertheless, further validation is required to determine the value of CABLES1 and CDKN1B immunostaining as a marker of CD since they have only been investigated in a small number of patients from a single cohort.
GENETIC DEFECTS
Somatic USP8 hotspot variants
Somatic variants in a hotspot that encodes residues 718-720 of the USP8 protein are the most common genetic cause of CD, being found in 21-62% of patients, and are significantly more frequent in women than in men and in adults and teenagers than in children90,98,99,112-126. The clinical relevance of this genetic defect is unclear. USP8 variants were associated with smaller tumors in some studies99,112,114, but to larger tumors in other studies113,120,122,124. Another reported association is with higher UFC117, which in one study occurred together with higher serum cortisol suppression with low-dose dexamethasone113. Other studies have identified an enhanced response of circulating cortisol and ACTH with the 8 mg dexamethasone suppression test and the desmopressin stimulation test112,127. Additional research has linked USP8 with lower circulating ACTH91,99. Two groups found increased remission rates in cases with USP8 variants99,120. Others identified higher recurrence rates91,116,117,124 and/or earlier recurrences112,117.
The histopathological phenotype of corticotropinomas carrying somatic USP8 variants is not well defined either, although increased immunoreactivity for SST5 and MGMT point toward higher pharmacologically responsiveness98,99. Indeed, primary cultures from corticotropinomas carrying a specific USP8 variant display an increased response to the treatment with pasireotide128. In contrast, CDKN1B immunoreactivity is reduced, while at the mRNA level, there are reduced CDKN1B and increased CDC25A and MAPK4 expression, which could translate into an enhanced proliferative potential119,121. USP8 has been proposed as a pharmacological target since its in vitro inhibition leads to reduced proliferation and ACTH secretion in murine corticotropinoma-derived cells129.
COMBINATION OF MULTIPLE DISEASE MARKERS
Perhaps except for postsurgical hypocortisolemia, none of the proposed markers have been fully validated as a reliable predictor of clinical behavior and response to treatment. Nevertheless, the combination of multiple markers could achieve good predictive value. Along these lines, a recent study evaluated multiple markers in parallel, finding that clinical variables, such as tumor size and postsurgical cortisol, in combination with the expression levels of SSTR1 and CRHR1 mRNA, could reliably predict the clinical evolution and the probability of remission in patients with CD130. This strategy, however, requires the collection of fresh tumoral tissue for RNA extraction, which is used in reverse transcription and quantitative polymerase chain reaction. Given that most corticotropinomas are microadenomas, the collection of fresh tissue for research is often difficult. On the other hand, a selection bias is likely in RNA-based studies, given that they could only analyze cases with enough available tissue for genetic tests.
Other studies have evaluated disease severity scores based on clinical and biochemical parameters as predictors of disease outcomes, but most of them are not specific for CD. One CD-specific severity score has been validated in pediatric patients, considering the degree of hypercortisolemia, impaired glucose tolerance, hypertension, height Z score, body mass index Z score, time to diagnosis, and tumor size and invasion. This score identified racial disparities consisting on a more severe disease in Hispanic and African-American children. In concordance, Hispanic and African-American patients displayed a shorter time in remission compared to non-Hispanic White patients in the same study131.
CONCLUSION
The clinical behavior of CD is highly variable and involves complex diagnostic studies, a combination of multiple therapeutic strategies, long-term management of comorbidities, and a lifelong risk for recurrence. The identification of practical, robust, and reliable markers of clinical behavior would be of great help in guiding the clinical management of these individuals. Multiple possible markers have been proposed; however, most of them require additional evaluation in studies in large cohorts designed ad hoc. Further, translational research is required to identify and validate novel biomarkers for their clinical implementation in the short term.

