











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
In March 2020, the Word Health Organization declared the novel coronavirus disease (COVID-19) a global pandemic1. The presentation of COVID-19 varies considerably and includes asymptomatic individuals, patients with mild respiratory symptoms, and the more severe disease spectrum in patients requiring hospitalization. The most severe cases are complicated by multiple organ dysfunction syndrome (MODS), with the kidney being one of the most affected organs, and reflecting the entire spectrum of acute kidney injury (AKI)2,3. We, herein, focus on describing all aspects of COVID-19-associated AKI (C19-AKI), including its epidemiology, proposed pathogenesis, management, and long-term outcomes.
EPIDEMIOLOGY OF C19-AKI
Initial reports suggested that AKI, as defined by the kidney disease improving global outcomes (KDIGO) criteria4, was uncommon following acute COVID-19 infection. Subsequent observations showed that C19-AKI occurs in 20% to 50% of hospitalized cases, with variations per institution, and with a decreasing trend in its incidence as the pandemic has evolved5,6. AKI rates are higher than those observed in the Influenza H1N1 pandemic and other illnesses, suggesting a kidney-specific susceptibility to this infection7.
AKI studies have classified it according to the site of acquisition, whether detected in the community on patient admission, or if it develops during hospitalization. This is relevant given the different characteristics of these patients8. In Mexican patients with SARS-CoV-2 infection who developed AKI, approximately two-thirds of AKI were community-acquired C19-AKI, and one-third was hospital-acquired C19-AKI, and both scenarios led to a dismal prognosis8.
As expected, rates of C19-AKI are higher in patients admitted to an intensive care unit (ICU), with reported incidences of 50-90%, and mostly in patients on invasive mechanical ventilation (IMV)9. In this context, 30-60% of C19-AKI are detected in severe AKI stages (Stages 2 and 3 of the KDIGO classification), and 6% to 39% require organ support with renal replacement therapy (RRT)10,11.
As in other diseases, the development of AKI is a poor prognostic factor. C19-AKI has been associated with higher in-hospital mortality rates (C19-AKI stages 1-3, HR = 5.6 [95% CI: 5.0-6.3], and C19-AKI with RRT (HR = 11.3 [95% CI: 9.6-13.1]), longer hospitalizations (patients with C19-AKI, 11.6 [IQR: 6.9-20.9] days versus patients without AKI, 5.2 [IQR: 3.0-8.6] days)11,12), and an increased risk of developing chronic kidney disease (CKD) in survivors at 3 months; 16% of all AKI survivors had CKD12,13.
RISK FACTORS FOR C19-AKI
There are many risk factors linked with the development of C19-AKI in clinical studies that vary depending on the population selection, differences in AKI prevalence, the AKI criteria applied, and the timing of hospital admission, among others. For example, C19-AKI is more commonly reported in studies conducted in countries other than China, or when based on a prospective clinical design instead of a retrospective analysis14. However, despite the heterogeneity of clinical studies, the most consistent risk factors found to be strongly associated to the development of C19-AKI are COVID-19 severity, older age, male sex, and a previous CKD diagnosis (Table 1).
Table 1. Risk factors associated with C19-AKI (adapted from Cai et al.14 and Raina et al.15)
| Risk factor | Association strength |
|---|---|
| Non-modifiable risk factor | |
| Elderly | OR = 3.94 (95% CI: 1.55–9.99) for age ≥ 60 years |
| Male sex | OR = 1.37 (95% CI: 1.25–1.49) |
| Non-Asian | OR = 2.34 (95% CI: 1.50–3.64) |
| Risk factors related to baseline health status | |
| Previous CKD | OR = 4.56 (95% CI: 3.63–5.73) |
| CAD | OR = 1.98 (1.74–2.24) |
| Hypertension | OR = 1.85 (95% CI: 1.70–2.02) |
| DM | OR = 1.71 (95% CI: 1.59–1.84) |
| Lung disease | OR = 1.36 (95% CI: 1.16–1.60) |
| Cancer | OR = 1.26 (95% CI: 1.13–1.40) |
| Risk factors related to severe COVID-19 | |
| Mechanical ventilation | OR = 8.61 (95% CI: 5.63–13.17) |
| Use of vasopressors | OR = 8.33 (95% CI: 4.72–14.72) |
| Risk factors, potentially modifiable | |
| Smoking | OR = 1.23 (95% CI: 1.07–1.42) |
| BMI > 30 kg/m2 | OR = 1.12 (95% CI: 1.01–1.25) Borderline risk, in a few cohorts the association is only observed when BMI > 35 kg/m2 |
OR: odds ratio; cardiovascular disease (CAD): coronary artery disease and heart failure; DM: diabetes mellitus; BMI: body mass index.
Undoubtedly, the key factor for C19-AKI is the severity of COVID-1914. Severe COVID-19 is directly associated with ICU admission, the development of acute respiratory distress syndrome (ARDS), IMV, the use of vasopressors, and MODS; all are strongly linked with the development of C19-AKI15. Likewise, many surrogates of severe inflammation caused by COVID-19 such as high levels of ferritin, D-dimer, lactate dehydrogenase, IL-6, or creatine kinase have been associated with C19-AKI8. The risk of C19-AKI in symptomatically mild COVID-19 seems odd, but there are case series reports of patients with mild COVID-19 and severe kidney dysfunction and/or heavy proteinuria15,16. The risk factors for C19-AKI identified in this population are the same as those reported in patients with severe COVID-19, such as male sex, older age, obesity, hypertension, diabetes, and pre-existing CKD, among others15. In several series, cases of severe AKI and proteinuria in patients with mild COVID-19 have been identified in three contexts: relapses of previous glomerular disease, de novo glomerulopathies, and those associated with SARS-COV-2 vaccination. These populations harbor risk factors such as pre-existing kidney disease, high-risk APOL1 genotypes, molecular mimicry of an antigen in individuals with an underlying genetic susceptibility, or specific HLA haplotypes16,17.
In contrast, interventions that decrease the risk of severe COVID-19 such as vaccination or dexamethasone administration effectively decrease the number of C19-AKI cases18. These “protective factors” could possibly explain the declining rates of C19-AKI throughout the pandemic19. Another possible, but rarely mentioned risk factor for C19-AKI, is the virulence of the variant sub-type of the SARS-CoV-2 virus. Studies have indicated that the risk posed by the omicron variant (B.1.1.529) in terms of hospitalization is lower than that of the delta variant (B.1.617.2)20. It is plausible that the decreased severity of COVID-19 due to the omicron variant translates into a lower rate of C19-AKI. Other risk factors are shown in table 1.
However, many risk factors remain inconclusive. Transplantation is considered a potential risk factor for AKI in many infectious diseases, given the high prevalence of pre-existing limited graft function and comorbidities, and the possible detrimental effects of immunosuppressive drugs in kidney transplant recipients (KTR)21. In a recent systematic review of 5,559 KTR, the risk of C19-AKI was higher in these patients compared with non-transplanted patients, regardless of sex, age, and comorbidities22. Even so, less biased larger studies revealed a similar rate of AKI in KTR in comparison with non-transplanted COVID-19 patients22. Given the broad heterogeneity of KTR characteristics, adjusting the effect for important confounders is difficult to achieve.
The use of certain medications before hospital admission may be a relevant risk factor for COVID-19 outcomes. Several meta-analyses have demonstrated that the use of renin-angiotensin-aldosterone system inhibitors (RAASis) does not have a detrimental effect on the clinical course of SARS-CoV-2 infection23,24. In a systematic review, a medical history of RAASi use in hospitalized patients was associated with a moderate to a severe increase in the risk of C19-AKI (OR = 1.68, 95% CI: 1.19-2.36); however, only retrospective studies were included in this review25. Contrarily, a randomized controlled trial found that the risk of severe AKI requiring hemodialysis was not different in COVID-19 hospitalized subjects who continued receiving RAASi compared with those who discontinued the medication26.
Other “prognostic” factors associated with the course and outcome of renal injury are rarely reported. Risk factors should be reported, as recommended, according to the biphasic pattern of C19-AKI: “early” AKI (AKI manifested at hospital admission) and “delayed” AKI (AKI developed during hospitalization)8. Further, there is a need to recognize and identify modifiable and preventable C19-AKI risk factors.
PATHOPHYSIOLOGY OF C19-AKI
The pathophysiological mechanisms of C19-AKI are multifactorial and include systemic immune and inflammatory responses induced by the viral infection, endothelial injury, and direct epithelial infection2. Furthermore, patients with COVID-19 usually present with volume depletion resulting from fever, shortness of breath, and gastrointestinal losses (Fig. 1)27. These findings are summarized in the following subsections.
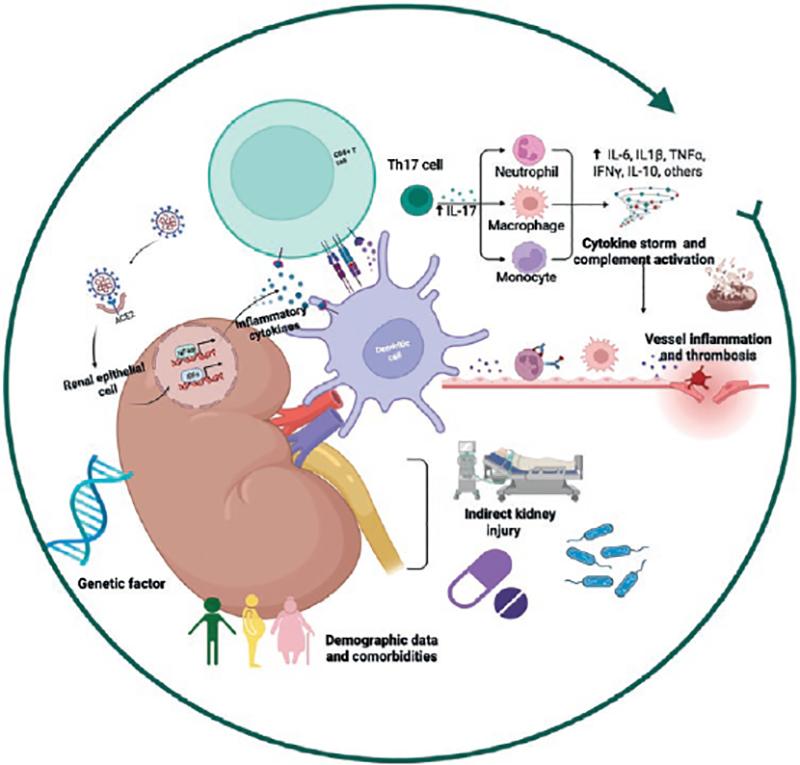
Figure 1. Pathophysiological mechanisms of C19-AKI.
The pathophysiologic
mechanisms of C19-AKI are multifactorial and include systemic immune and
inflammatory responses induced by viral infection, endothelial injury, and
direct epithelial infection. On the other hand, several non-specific
mechanisms may contribute to the development of C19-AKI, including
hemodynamic abnormalities, hypovolemia from poor oral intake or high fever,
use of nephrotoxic drugs, and hospital-acquired sepsis. Risk factors for
C19-AKI, sex, age, and hereditary susceptibility, are of relevance in the
pathogenesis.
Viral tropism for the kidney
The SARS-CoV-2 virus uses the angiotensin-converting enzyme 2 (ACE2) receptor as a gateway to infect human cells28. ACE2 RNA is expressed in multiple organs, such as the lung, intestines, and kidney. However, ACE2 expression is greater in the kidney than in the lung; the main cells expressing this enzyme in the kidney are podocytes and proximal tubular epithelial cells and are, hence, more susceptible to infection and subsequent injury27,29-31.
Most ACE2 is present in insoluble form and bound to cell membranes. ACE2, in both the soluble and insoluble forms, converts angiotensin II to angiotensin (1–7), which is crucial to the control of different functions in the body2,28. The SARS-Cov-2 viral spike glycoprotein also binds with high affinity to ACE2, triggering its internalization along with the virus28,31. Intracellular translocation of SARS-CoV-2 and ACE2 leads to its depletion, loss of its normal endogenous enzyme function, and concomitant depletion of its catalytic product, Ang (1-7), thus hindering its vasodilatory effects, antiproliferative, and antifibrotic functions32,33.
Although these are possible explanations, there is no convincing evidence of direct SARS-CoV-2 renal tropism. Few studies have been able to demonstrate the presence of viral particles in renal tissue, and many have failed to do so16,34,35. These discrepancies could result from the time lapse between infection and biopsy procurement, the severity of the condition, and the samples’ processing techniques36. On the other hand, various reports documenting SARS-CoV-2 virions or virus-like particles also describe physiological structures and organelles, such as clathrin-coated vesicles, multivesicular bodies, rough endoplasmic reticulum, and unidentified subcellular structures. This problem results primarily from the techniques used in its detection37.
Tubular and glomerular injury
Kidney injury caused by COVID-19 typically manifests as tubular damage with urinalysis abnormalities (low-grade proteinuria, hematuria, and/or impaired glomerular filtration rates)38. Renal biopsies and post-mortem studies frequently document acute tubular injury by light microscopy, and in some reports, viral particles have been detected within renal tubular epithelial cells and podocytes by electron microscopy39. This last finding has been debated since they have not been documented in all biopsies, perhaps as a result of the processing technique, the severity of the disease, and the time lapse between infection and sample collection.
Many studies have reported patients with COVID-19 and a collapsing glomerular disease40,41. The two possible underlying mechanisms could be cytokine-mediated injury to podocytes and/or direct toxic viral effects on podocytes. Genetic susceptibility, especially the presence of high-risk apolipoprotein 1 (APOL1) genotypes, may play a key role in its pathogenesis40,41. Other de novo glomerulopathies or relapses of a previous disease have been subsequently reported42,43.
Complement activation
The complement system, a significant component of native immunity, is critical to the rapid response against infections. Due to its central role in the detection and removal of pathogens, complement should intuitively be protective in SARS-CoV-2 infection44. SARS-CoV-2 per se can activate the complement system directly through the lectin pathway, the classical pathway, and/or the alternative pathway45,46. Specifically, the spike and nucleocapsid proteins of SARS-CoV-2 can be directly recognized through the lectin pathway; IgG and IgM antibodies directed against the receptor-binding domain within the spike protein initiate activation of the classical pathway, and the SARS-CoV-2 spike protein can directly dysregulate the alternative complement activation pathway by binding to heparan sulfate and competing with factor H47,48.
Multiple lines of evidence implicate hyperactivation of the complement system in the pathophysiology of COVID-19. How and why the normally protective complement system becomes pathogenic in COVID-19 remains unknown44,45. Complement activation has been demonstrated in the kidney, with evidence of complement factor deposition and of the membrane attack complex in nephron vessels and on the tubular basement membrane46. These findings have led to studies on the effects of the complement C5a inhibitor, eculizumab, in COVID-19 patients; although preliminary results appear promising, these studies have not reached a sufficient number of patients to demonstrate any significant effects on C19-AKI49.
Cytokine activation
SARS-CoV-2 binding to ACE-2 receptors and viral replication in lung cells lead to epithelial and endothelial cell apoptosis and vascular leakage and promote the activation of the inflammatory cascade2. These inflammatory cytokines and chemokines then recruit more innate immune cells and activate adaptive immune cells to sustainably release more inflammatory cytokines that further aggravate the injury50.
Many studies have shown that COVID-19 patients have elevated levels of numerous inflammatory cytokines, including IL-1β, IL-2, IL-6, IL-10, IFN-γ, and TNF-α, and that these cytokines correlate with disease severity and outcomes51. Th17 cells have both a protective and a pathogenic role in COVID-19; they play an important role in its pathogenesis, not only activating the cytokine cascade but also inducing Th2 responses that inhibit Th1 differentiation and suppress Treg cells52.
Targeted immunomodulation in the management of COVID-19 infection (e.g., tocilizumab) and non-specific immunosuppression (e.g., corticosteroids) can be used to limit the collateral damage caused by the adversely activated immune system. Pharmacological treatments and extracorporeal blood purification therapies have been tested in an attempt to decrease circulating interleukin levels (IFN-γ, IL-16, and IL-10); however, this decrease in cytokines has not translated into an improvement in mortality53-55.
On the other hand, some observational studies have described that low levels of IL-4, IL-17, and IFN-γ herald a worse prognosis, but these results are controversial, because they depend on the timing of the sample collection, the site of procurement (BAL vs. serum), and its subsequent processing56.
COVID-19-associated coagulopathy
Endothelial cells contribute to the preservation of normal hemostasis by maintaining vessel wall integrity and balancing fibrinolysis through the expression of coagulation inhibitors and blood clotting enzymes57. SARS-CoV-2 may alter vascular homeostasis by directly infecting endothelial cells through ACE2. Furthermore, a decrease in ACE2 expression might, in turn, indirectly activate the kallikrein–kinin system, ultimately leading to increased vascular permeability2. Pro-inflammatory cytokines can also induce a capillary leak syndrome and thrombosis, which may result in disseminated intravascular coagulation2,27. The main hematological abnormalities reported in these patients consisted of leukopenia, lymphopenia, anemia, and thrombocytopenia, and the proportion of their decrease was usually a harbinger of disease progression and severity58,59.
The prothrombotic nature of COVID-19-associated sepsis has been well described. Platelet-rich thrombi have been observed in the microvasculature of the heart, brain, kidney, and liver; renal infarctions secondary to arterial thrombi have also been described, especially in the early phases of the pandemic57.
Indirect kidney injury
Several non-specific mechanisms may contribute to the development of C19-AKI, including hemodynamic abnormalities, cardiac dysfunction, hypovolemia due to poor oral intake or high fever, the use of nephrotoxic drugs (antibiotics or contrast medium), and hospital-acquired sepsis57. Furthermore, the high positive-pressure levels used in IMV can potentially induce or exacerbate abnormalities in intrarenal blood flow and promote other hemodynamic changes that impair renal perfusion2,57. All these direct and indirect mechanisms, some of them speculative, contribute to the development of C19-AKI (Fig. 1).
C19-AKI: RECOGNITION
Early diagnosis of AKI was a challenge during the COVID-19 pandemic and in every discipline of healthcare. In Mexico, renal involvement at hospital admission was more prevalent in older patients8, and in those with a history or associated comorbidities (hypertension, CKD, and obesity), conditions characteristically associated with decreased renal reserves8,60,61.
The precise etiology of C19-AKI in different cohorts, including Mexican cohorts, cannot yet be fully elucidated, and given the diversity in its presentation; in many cases, the etiology is multifactorial. However, sepsis and hemodynamic instability may play a predominant role in these cases5,8. The timing between the initial development of AKI and hospital admission varies between cohorts. In a multicentric study, Hirsch et al.5 described that most cases developed early, with 37.3% of cases manifesting AKI at admission or developing it within 24 h of admission; they also observed a significant relationship between the presence of respiratory failure and AKI, unmistakable manifestations of the inflammatory phase of the disease5,61.
In 2020, the 25th Consensus Conference of the acute disease quality initiative (ADQI) focused on this new disease3. They recommended the use of the KDIGO criteria to define and report C19-AKI and thus enable reproducibility in study comparisons, and improve survival with the increased and timely recognition of this entity4. Traditionally, an AKI diagnosis is based on changes in kidney function such as increased serum creatinine (sCr), which is a low sensitivity method since nearly 50% of the glomerular filtration rate (GFR) must be lost before a change in sCr is detectable, and decreased urine output, which lacks specificity since it may be triggered by hypovolemia and totally unrelated with kidney injury3.
The severity of C19-AKI may be classified with the well-documented consensus KDIGO criteria for AKI staging, and outcomes appear to correlate with the severity of AKI as defined by this classification system4. As mentioned above, the severity of C19-AKI may lead to different outcomes. In addition, the concept of the duration of AKI should also be considered in the description of AKI, as recently described in a prospective cohort by Casas et al., in which persistent AKI was also associated with increased mortality (HR = 7.42, 95% CI: 1.04-53.04, p = 0.046)62.
Additional tools may be useful to confirm or support a C19-AKI diagnosis and to establish differential diagnoses and prognostic assessments4. Urinalysis and urine chemistries such as urinary sodium (UNa) levels, fractional excretion of sodium (FeNa), and fractional excretion of urea (FeU) are commonly used to differentiate pre-renal AKI from acute tubular necrosis (ATN), but they remain insensitive and non-specific markers, offering little reliable information to establish a diagnosis of AKI. Martínez et al.8, analyzed the urinalyses and urine electrolytes of patients with community-acquired and hospital-acquired C19-AKI, and they reported a high prevalence of FeNa < 0.5% (71 vs. 71%), a similar proportion of cases with proteinuria > 2 + (34 vs. 41%), but a higher proportion of hematuria in patients with hospital-acquired C19-AKI (59 vs. 28%). These findings are in line with a study reported by Morita et al.63, in which the urinary sediment findings in subjects with COVID-19 were more severe as renal function declined. These two cohorts suggest that glomerular hypoperfusion might be largely involved in the pathogenesis of C19-AKI and at least partially conserved tubular sodium absorption, without a full-blown classic acute tubular injury presentation8,63. Thus, the use of these markers in C19-AKI appears to be even more limited since C19-AKI has a complex pathophysiology that affects more functions other than tubular reabsorption.
Biomarkers can help to differentiate various types of kidney injury and may be uniquely helpful in quantifying tubular injury in COVID-19 and predicting progression of AKI stage, the need for RRT, or death62. The elevation of kidney injury urinary biomarkers in the absence of changes in sCr and a normal urine output have been considered subclinical AKI, a term identifying those patients at high risk of AKI. These patients are likely to benefit from obtaining biomarkers and performing early interventions. In this context, the tissue inhibitor of metalloproteinases-2 (TIMP-2) and the insulin-like growth factor binding protein-7 (IGFBP7) have been identified as possible AKI biomarkers, given that both are released following ischemic or inflammatory processes in the kidney, resulting in transient G1 cell cycle arrest64. Another biomarker of early AKI is the urinary neutrophil gelatinase-associated lipocalin (NGAL); the intrarenal concentration of this protein is abruptly up-regulated soon after ischemic or nephrotoxic kidney injury64. A recent Mexican cohort study of critically ill patients with SARS-CoV-2 infection and without functional criteria of AKI established that the result of the formula (TIMP-2) × (IGFBP7) > 0.2 (ng/mL)2/1000 was associated with AKI during hospitalization (HR = 7.23 [0.99-52.4], p = 0.050)62. These biomarkers, in conjunction with clinical findings, were useful to identify subclinical AKI in critically ill COVID-19 patients. Table 2 summarizes the different urinary biomarkers used in hospitalized patients with C19-AKI62,65-71.
Table 2. Feature selection and machine learning models performance in training and testing stages, for each of the COVID-19 groups according to the basic and extended profile
| Basic profile | Extended profile | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Features | AUC (95%CI) | Sensitivity | Specificity | Accuracy | Features | AUC (95%CI) | Sensitivity | Specificity | Accuracy | |
| COVID-19 versus Non-COVID-19 |
Cough | Training | Cough | Training | ||||||
| Obesity | 0.95 (0.82–0.96) |
0.90 | 0.92 | 0.91 | Trimethylamine N-oxide | 1 (1–1) |
0.86 | 0.97 | 1 | |
| Fever | Lyso PC a C 26:0 | |||||||||
| Dysgeusia | Lyso PC a C 18:2 | |||||||||
| Anosmia | Blind Test 0.88 (0.74–1) |
1 | 0.91 | 0.93 | IFN-α2 PC aa C 36:6 C 10:1 |
Blind Test 0.93 (0.81–1) |
1 | 0.95 | 0.96 | |
| Mild COVID-19 versus Severe and Critical COVID-19 |
Tiredness | Training | C 10:2 | Training | ||||||
| Neutrophils | 0.96 (0.88–0.98) |
0.94 | 0.92 | 0.93 | Neutrophils | 0.98 (0.96–1) |
1 | 0.89 | 0.92 | |
| Monocytes | Lyso PC a C 14:0 | |||||||||
| Lymphocytes | Dyspnea | |||||||||
| Chills | Lymphocytes | |||||||||
| Cough | Blind Test | 0.92 | 0.60 | 0.78 | Methionine sulfoxide | Blind Test | 1 | 0.92 | 0.95 | |
| Arthralgia | 0.80 (0.56–0.92) |
Neutrophils lymphocytes Ratio | 0.97 (0.85–1) |
|||||||
| Myalgia | Trimethylamine N-oxide | |||||||||
| Transhydroxyproline | ||||||||||
| Total dimethylarginine | ||||||||||
| Severe
COVID-19 versus Critical COVID-19 |
Training | Training | ||||||||
| Leukocytes | 0.91 (0.73-0.92) |
0.91 | 0.81 | 0.87 | 0.85 (0.94–1) |
0.88 | 0.75 | 0.82 | ||
| Creatinine | Trimethylamine N-oxide | |||||||||
| Vomit | p Hydroxyhippuric acid | |||||||||
| Rhinorrhea | Blind Test | 0.62 | 0.87 | 0.75 | Lyso PC a C 28:1 | Blind Test | 1 | 0.42 | 0.75 | |
| 0.67 (0.41–0.88) |
0.68 (0.47–0.92) |
|||||||||
| Survivors versus Non survivors |
Chills | |||||||||
| Vomit | ||||||||||
| Diarrhea | Training with 100% of data | SM OH C 16:1 | Training with 100% of data | |||||||
| Abdominal pain | 0.68 (0.61–0.76) |
0.50 | 0.86 | 0.73 | PC aa 32:2 | 0.62 (0.52–0.72) |
0.47 | 0.78 | 0.66 | |
| Sex | PC ae C 36:0 | |||||||||
| Smoking | ||||||||||
| Headache | ||||||||||
AUC: area under curve; uNGal: urinary neutrophil gelatinase-associated lipocalin; AKI: acute kidney injury; RRT: renal replacement therapy; NR: not registered; uKIM: urinary kidney injury molecule-1; HR: hazards ratio; TIMP-2: tissue inhibitor of metalloproteinase type 2; IGFBP7: Insulin-like growth factor binding protein; EGF: epidermal growth factor.
MANAGEMENT OF C19-AKI
According to the KDIGO guidelines, the management of patients with C19-AKI should not differ from that of patients with AKI due to other causes4. However, there are some particularities in COVID-19 that should be considered (Fig. 2).

Figure 2. Management of C19-AKI. Comprehensive management of the patient with C19-AKI includes optimal fluid management, hemodynamic monitoring, glucose control, anticoagulation, nutrition, and immunomodulatory agents, as discussed in the text.
Optimal fluid management is essential. In C19-AKI, we have seen dynamic courses in which hypovolemia secondary to fever, cough, diarrhea, anorexia, and hypercatabolism can develop during early infection. Once hospitalized, some patients require intubation, which further promotes volume depletion. Appropriate fluid resuscitation and avoiding fluid overload are essential steps at this stage, and balanced crystalloids should be considered as first-line treatment72. On the other hand, during hospitalization, it is not uncommon that patients become fluid overloaded72. Therefore, objective and dynamic evaluation of intravascular volume and heart and lung status with conventional point-of-care ultrasonography (POCUS) could be very useful72. In the presence of shock, early initiation of vasopressors is necessary in AKI, and it seems reasonable to apply this approach within the context of COVID-1972.
In patients taking RAASi, these should not be discontinued unless strictly necessary26. Interestingly, based on the pathophysiological mechanism of the entry of SARS-Cov-2 into cells, in a multicenter clinical trial with early COVID-19 patients who were randomized to receive telmisartan 40 mg or placebo every 12 h for 2 weeks, C-reactive protein decreased in cases who received telmisartan; they also had a shorter hospital stay, as well as decreased mortality73. This information is in line with results from other AKI cohorts, in which the administration of RAASi during or as early as possible after hospitalization improves clinical outcomes.
Other general measures recommended in AKI due to any cause and that are recommended in C19-AKI include avoidance of drugs with nephrotoxic potential (analgesics or aminoglycosides), if possible, avoiding hyperglycemia, and the use of immunomodulatory agents. In hospitalized patients with C19-AKI, hyperglycemia may be secondary to insulin resistance, a hypercatabolic state, and/or the result of dexamethasone (DXM) administration. Glucose levels should be maintained ~140-180 mg/dL and, if necessary, the use of insulin in bolus or infusion is recommended74,75.
Immunomodulatory agents that may potentially attenuate cytokine production such as tocilizumab, sarilumab, anakinra, imatinib, dasatinib, cyclosporin, immunoglobulins, or baricitinib have no demonstrated benefit or effect on the prevention or amelioration of C19-AKI3. Baricitinib is one of the most often used drugs: we must recall that its half-life may extend up to 19 h (normally, 6 h) in patients with severe AKI, and it is rapidly dialyzable3. Remdesivir, an inhibitor of viral RNA polymerase, can improve C19-AKI by modulating macrophage inflammasome activation and the inflammatory immune response76. In mice, the drug protects against AKI by specifically inhibiting the NLRP3 inflammasome activation in macrophages, thereby reducing inflammation, and improving the recovery of renal function and the kidney pathological score; however, these effects have not been proven in humans76. Further, there is evidence that remdesivir may be associated with the development of AKI, increasing the risk of kidney injury 20-fold. To date, renal function in patients on remdesivir should be closely monitored, and its use should be avoided if the eGFR is < 30 mL/min76.
In the RECOVERY trial77, DXM treatment improved survival at 28 days in the subgroup of patients with severe (patients with oxygen requirements) and critical (ICU patients) COVID-1948. Interestingly, in this study, a lower percentage of patients warranted RRT (4.4% vs. 7.5%, OR = 0.61) if they had received DXM, although renal outcomes were not one of the study’s main objectives77. Subsequently, in a cohort study of 100 patients with severe COVID-19 conducted by another group, they reported that the systematic use of DXM prevented the development of AKI (75% vs. 35%, OR = 0.31; CI: 0.09–0.99)77.
Anticoagulation has become essential in the treatment of hospitalized patients with COVID-19. AKI is considered to carry a high risk of thrombosis, and prophylactic anticoagulation should be administered. Some anticoagulants should be adjusted to renal function such as low molecular weight heparins (enoxaparin) whose dose should be decreased by half, but not that of unfractionated heparin. In the absence of thrombosis, anticoagulation should be continued for 4 weeks with direct anticoagulants (apixaban and rivaroxaban, which do not require renal dose adjustment), and for 6 months if thrombosis is confirmed during hospitalization78.
The nutrition of patients with C19-AKI who do not require RRT should guarantee a protein intake of 1.3-1.5 g/kg/day, and it should be increased if hemodialysis (HD) or continuous kidney replacement therapy (CKRT) are required3.
In patients with C19-AKI, kidney injury appears to develop as a result of renal ischemia and acute tubular necrosis with an acquired renal deficiency in intermediate energy metabolism (NAD+: nicotinamide adenine dinucleotide); a deficiency or shortage of this decreases ATP production that, if not controlled, can culminate in tubular dysfunction and cell death80. Based on these findings, a cohort study reported that patients who received oral niacinamide (Vitamin B3) had a 36% decrease in their risk of death and the need for RRT, especially in patients with a greater severity score in the KDIGO 2-3 criteria80.
In kidney-transplanted patients with C19-AKI, treatment with bamlanivimab or casirivimab-imdevimab and immunosuppression reduction has been tested with some success. Patients treated with monoclonal antibodies developed measurable anti-SARS-CoV-2 IgG with ACE2 receptor blocking activity, and no reported associated deaths or graft losses81.
RENAL REPLACEMENT THERAPY
In patients with C19-AKI, the indications to initiate RRT remain the same as in patients with AKI unrelated to COVID-19. However, in the early waves of the pandemic, when the number of patients was very high, the approach of waiting as long as possible before resorting to RRT was the most common practice82.
The options for RRT in C19-AKI patients mainly depend on the center, the availability of the necessary equipment, and the expertise of the medical and nursing staff for the management of different therapies. RRT modalities are divided into continuous RRT (CRRT), prolonged intermittent RRT (PIRRT), intermittent hemodialysis (IHD), and peritoneal dialysis (PD)83 (Fig. 3).

Figure 3. Renal replacement therapy (RRT). The options for RRT in C19-AKI are divided into continuous RRT, prolonged intermittent RRT, intermittent hemodialysis, and peritoneal dialysis.
Some cohort studies have recommended CRRT as the first-choice therapy since it leads to better hemodynamic control in patients that in most cases, were hemodynamically unstable, on IMV, requiring multiple vasopressors, and with an overexpressed inflammatory response. However, many centers did not have CRRT available, or not a sufficient amount to care for all patients in need. Therefore, PIRRT was used instead, and it became a good option, with favorable results83. On the other hand, hemodynamically stable patients or those with emergency indications, such as hyperkalemia or severe metabolic acidosis, were considered for IHD treatment. During this pandemic, PD became a cornerstone of therapy, and proved to be a good option in hemodynamically unstable patients, particularly in those with small-volume refills (1.5 L), as proposed by Ponce et al.84. A controversial issue that arose with PD was its use in patients requiring a prone position. However, with these small intra-peritoneal volumes, the patient position did not appear to be a problem, although, according to the world literature, this modality of treatment had been reserved for patients in the supine position84.
EXTRACORPOREAL BLOOD PURIFICATION
Patients with severe COVID-19 develop AKI as part of the broad spectrum of MODS, so it was not uncommon for them to require multi-organ support, better known as extracorporeal organ support (ECOS)85. ECOS has become a reality and a useful weapon against COVID-19 and its complications. In an attempt to remove inflammatory mediators such as Pathogen-Associated Molecular Patterns (PAMPS), Damage-Associated Molecular Patterns (DAMPS), cytokines, and endotoxins, the use of sequential extracorporeal therapies has become more frequent than ever85. Table 3 shows some of the most-used extracorporeal therapies and their rationale. Although these treatment modalities have not reaped major survival benefits, and there is no clear evidence of their benefits in randomized clinical trials, their underlying physiological bases are promising in patients with sepsis and COVID-1985.
Table 3. Blood purification techniques in COVID-19 patients
| Technique | Mechanism and examples |
|---|---|
| HVHF and HCO | HVHF: Convection enhances removal of large molecular proinflammatory cytokines |
| HCO: Large-pore membrane (average 20 nm) allows removal of large cytokine molecules | |
| Hemadsorption and Hemoperfusion (HP) | Membranes coated with heparin or antibiotics. Adsorption of endotoxins and cytokines (Oxiris, Septex) |
| HP involves passage of blood through a hemofilter where mediators are adsorbed to the membrane surface or through a sorbent-containing cartridge | |
| (Cytosorb, Toraymyxin, HA330, CPFA): Adsorption of endotoxins and cytokines. (Seraph): Bacteria, viruses, fungi, and toxins | |
| TPE | An extracorporeal technique performed in an apheresis machine in which patient plasma is separated from blood and removed; then, the cellular blood components are returned to the patient together with a replacement fluid |
HVHF: high-volume hemofiltration; HCO: high cut-off membrane; HP: hemoperfusion; TPE: therapeutic Plasma Exchange; ECCO2R: extracorporeal carbon dioxide removal; ECMO: extracorporeal membrane oxygenation.
LONG-TERM OUTCOMES IN C19-AKI
According to observational studies, 30-80% of patients with C19-AKI reach different grades of kidney function recovery at follow-up11,13,86, although little is known about long-term kidney function at 1 year and beyond. In the multicenter cohort study, STOP COVID, of 637 patients admitted to the ICU with AKI and RRT, 63% had died at the end of a median follow-up of 17 days. Of the 216 patients discharged, 34% were still dialysis-dependent at discharge, and 18% at 60 days after ICU admission12. Another cohort that exclusively included critically ill patients with C19-AKI showed a 74% recovery of kidney function at discharge in cases with AKI without the need for RRT, and 67% in those with AKI and RRT13. An even better renal prognosis was found in a retrospective study of patients admitted to the ICU in an U.K. tertiary center, where 82% of patients had full renal recovery at discharge, and 91% recovered at 90 days, defining recovery as sCr < 1.5 times of baseline and no need for RRT88. This broad range of outcomes can be explained in part by the heterogeneity of the studied population, different follow-up periods, and the lack of a standardized definition of kidney recovery.
Notably, a cohort study from the United States revealed an accelerated decline in kidney function in patients with C19-AKI and non-RRT during hospitalization, and who survived after discharge. After a median follow-up of 93 days, their eGFR had decreased more than that of controls with AKI and no COVID-19, with a difference in slope of −11.3 mL/min/1.73 m2/year (95% CI; −22.1-−0.4; p = 0.04) in the unadjusted mixed-effects model; −12.4 mL/min/1.73 m2/year (95% CI, −23.7-−1.2; p = 0.03) after adjusting for comorbidities; and −14 mL/min/1.73 m2/year (95% CI, −25.1-−2.9; p = 0.01) in the adjusted model for comorbidities, peak SCr, and the need for dialysis86. This study underscores the relation between C19-AKI and subsequent CKD, as well as its association with a steeper decline in kidney function than in AKI due to other causes; these observations suggest worsening outcomes at follow-up and should alert us as clinicians to manage closely and strictly control CKD progression factors.
To date, the largest registry that has evaluated renal function outcomes after COVID-19 infection was conducted by Bowe et al.87. They studied 89,216 patients with COVID-19 and compared them to 1,637,467 controls. At 6 months, COVID-19 survivors had a greater risk of AKI (HR = 1.94 [CI 95%: 1.86-2.04]), their eGFR decreased ≥ 50% (HR = 1.62 [CI 95%: 1.51-1.74]), and their risk for end-stage kidney disease (ESKD) increased (HR = 2.96 [CI 95%: 2.49-3.51]), as did their risk of developing a major adverse kidney event (MAKE) (HR = 1.66 [CI 95%: 1.58-1.74]). The risk increased according to the severity of the disease, whereby non-hospitalized individuals (81.5% of the cohort) had the lowest risk, while hospitalized patients (14%) and those managed in an ICU (4.7%) showed the greatest risk of developing adverse kidney outcomes. When the authors classified patients according to AKI development, hospitalized patients with AKI had a greater risk of ESKD (HR = 4.91 [CI 95%: 3.10-7.77]) and MAKE (HR = 7.37 [CI 95%: 6.03-9.02]), in comparison with non-hospitalized COVID-19 patients. The same phenomenon was observed in terms of ESKD (HR = 3.37 [CI 95%: 1.88-6.06]) and MAKE risks (HR = 2.92 [CI 95%: 2.33-3.65]), in contrast with hospitalized COVID-19 individuals who did not develop AKI. Linear mixed models to characterize post-acute COVID-19 eGFR trajectories showed a greater decline in eGFR in patients with COVID-19 admitted to the ICU: −7.69 (−8.27-−7.12), −5.2 (−5.24-−4.16) in hospitalized cases, and −3.26 (−3.58-−2.94) in outpatients, compared with an average slope of −0.49 mL/min/1.73 m2/year in controls. Likewise, when patients were stratified by AKI development, a decline in eGFR – 8.41 (CI 95%: −9.72-−7.10) mL/min/1.73 m2/year was observed in hospitalized individuals with AKI, in comparison with a drop of −5.27 (CI 95%: −5.86-−4.68), and –3.30 (CI 95%: −3.62-−2.99) in cases that were hospitalized without AKI, and in non-hospitalized patients, respectively87. These authors demonstrated the relevance of COVID-19 infection in kidney outcomes during follow-up and adequately revealed how individuals with more severe disease develop more renal function sequelae. The value of this study hinges not only on its sample size but it is also the first study demonstrating that even patients without AKI during the course of COVID-19, have worse renal prognoses in terms of AKI, ESKD, eGFR decline, and MAKE.
In our experience at the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, in a registry of 60 patients with C19-AKI who required RRT, 29 patients were discharged alive (48%). At 90 days, no patients remained dialysis-dependent, 77% (n = 23) reached complete remission, and 20% (n = 6) had partial recovery of renal function. Twenty-six (90%) patients have completed their one-year follow-up evaluation. Four individuals (15%) have died and 3 (12%) as a result of urgent dialysis initiation criteria. Furthermore, 12% (n = 3) were diagnosed with advanced CKD stages 4-588. These findings are in line with the previous reports throughout the world.
CONCLUSIONS
Despite early reports on the contrary, AKI as a complication of COVID-19 infection is very common in hospitalized patients, and more so in ICU patients requiring IMV. The development of C19-AKI increases the risk of mortality, CKD, and ESRD. The treatment should follow practice guidelines for the general management of AKI given the heterogeneous nature of potential AKI causes. No specific therapies have demonstrated a clear benefit in patients with C19-AKI. In some studies, DXM decreased the need for RRT and the incidence of AKI. Extracorporeal blood therapies appear promising, but at this point it should be preferably tested within the context of a clinical trial. Long-term outcomes of C19-AKI are poor, and these patients should be closely followed considering their poor renal function prognosis.

