










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalud Pública de México
versión impresa ISSN 0036-3634
Salud pública Méx vol.46 no.6 Cuernavaca nov./dic. 2004
ARTÍCULO ESPECIAL
Ensayos clínicos aleatorizados: variantes, métodos de aleatorización, análisis, consideraciones éticas y regulación
Eduardo Lazcano-Ponce, MC, Dr en CI; Eduardo Salazar-Martínez, M en C, Dr en CI, II; Pedro Gutiérrez-Castrellón, MC, Dr en CIII; Angélica Angeles-Llerenas, MC, M en CI; Adolfo Hernández-Garduño, MC, M en CIV; José Luis Viramontes, MC, M en C.V
ICentro de Investigación en Salud Poblacional. Instituto Nacional de Salud Pública. Cuernavaca, Morelos, México
IIInstituto Mexicano del Seguro Social, Delegación Morelos. Cuernavaca, Morelos, México
IIIDepartamento de Metodología de la Investigación. Instituto Nacional de Pediatría. México, DF, México
IVServicio de Pediatría, Hospital General de México, Secretaría de Salud. México, DF, México
VInvestigador independiente. México, DF, México. México, DF, México
"Me levanté muy temprano para visitarlos, con la esperanza de encontrar a aquellos a quienes había administrado un medicamento digestivo. Sintiendo un poco de dolor, sus heridas no habían crecido o inflamado y habían podido dormir durante la noche. Los otros a quien yo había aplicado un aceite hirviendo tuvieron fiebre con mucho dolor y protuberancias alrededor de sus heridas. Entonces yo determiné nunca otra vez quemar así tan cruelmente a los pobres heridos por arquebus".1
Ambroise Paré (1510-1590).
Los estudios epidemiológicos son clasificados, se gún se asigna la exposición, en experimentales y observacionales. La característica principal de los estudios experimentales es que el investigador asigna en forma aleatoria a la exposición. En relación con la característica de temporalidad, estos estudios son de carácter prospectivo, y por el número de observaciones sucesivas realizadas durante el periodo de estudio, son catalogados como longitudinales. Asimismo, a diferencia de los diseños de investigación observacional, donde los criterios de selección de la población se basan en la presencia del desenlace (casos y controles) o exposición (cohorte), los estudios experimentales no tienen estos dos criterios de selección y, generalmente, incluyen poblaciones homogéneas que puedan ser comparables en cuanto a su condición de enfermedad y características biológicas y sociodemográficas. En estos estudios las unidades de análisis pueden ser individuales o grupales ("clusters" o intervenciones comunitarias).
El diseño experimental clásico tiene diferentes características que lo definen.2 La primera es el control de las condiciones bajo estudio, esto es, la selección de los sujetos, la manera como el tratamiento es administrado, la forma en la que las observaciones son obtenidas, los instrumentos usados para realizar las mediciones; los criterios de interpretación deben ser implementados lo más uniforme y homogéneamente posible. La segunda es que debe haber una maniobra de intervención bajo estudio y al menos un grupo control. Tercera, los participantes en el estudio deben ser asignados en forma aleatoria a los grupos de intervención; esto es, ningún investigador, clínicos participantes, o sujetos de estudio, deben participar por sí mismos en la decisión del tratamiento que ellos recibirán. La cuarta es que la población de sujetos de estudio debe estratificarse en subgrupos, por diversos factores, como edad, sexo, grupo étnico y severidad de la condición clínica estudiada; esto con el fin de restringir las comparaciones a los sujetos que forman parte del mismo subgrupo. Finalmente, entre otras posibles, la quinta es que en un diseño experimental se requiere que el evento de interés (outcome) sea perfectamente definido y cuantificado antes y después de haber recibido la intervención.3 Es necesario considerar que el término ensayo es equiparable en este tipo de estudios a experimento.
Definición de ensayo clínico controlado aleatorizado
Un ensayo clínico es un experimento controlado en voluntarios humanos que se utiliza para evaluar la seguridad y eficacia de tratamientos o intervenciones contra enfermedades y problemas de salud de cualquier tipo; así como para determinar efectos farmacológicos, farmacocinéticos o farmacodinámicos de nuevos productos terapéuticos, incluyendo el estudio de sus reacciones adversas. Esto es, un ensayo clínico es un experimento con pacientes como sujetos de estudio, en el cual cuando se prueba un nuevo medicamento se comparan al menos dos regímenes de tratamiento uno de los cuales es denominado como control. Existen dos tipos de controles, los pasivos (negativos) y los activos (positivos). Un control negativo utiliza placebo en un ensayo de agentes terapéuticos, lo que significa la inclusión de un producto inocuo, cuya preparación por sí misma es similar en presentación, tamaño, color, textura y sabor a la de la preparación activa. En algunos casos en los que se desee demostrar que la preparación es equivalente o superior al producto estándar existente, y para proteger a pacientes que necesitan medicación por prescripción médica, deberá ser empleado un control activo.4
Los ensayos clínicos controlados aleatorizados (ECCA) son considerados el paradigma de la investigación epidemiológica, porque son los diseños que más se acercan a un experimento por el control de las condiciones bajo estudio y porque pueden establecer relaciones causa-efecto si las siguientes estrategias se establecen eficientemente: a) asignación de la maniobra de intervención mediante mecanismos de aleatorización en sujetos con características homogéneas que permiten garantizar la comparabilidad de poblaciones; b) la utilización de un grupo control permite la comparación no sesgada de efectos de dos posibles tratamientos, el nuevo, habitual o placebo; c) el cegamiento de los grupos de tratamiento permite minimizar los posibles sesgos de información y posibilita la comparabilidad de información, y d) finalmente, la incorporación de las estrategias descritas previamente permiten la comparabilidad en el análisis (figura 1).

Clasificación de ensayos clínicos
Los ensayos clínicos se plantean en forma muy diversa, por esta razón es necesario establecer criterios de clasificación. Cuando se estudian por la estructura de tratamiento pueden ser agrupados en diseños paralelos, de tratamiento sucesivo y ensayos alternativos. En relación con el enfoque de enfermedad, adicional a los ensayos de tratamientos terapéuticos, se pueden poner en práctica ensayos de prevención primaria y secundaria. En cuanto al enfoque de tratamiento, los ECCA estudian efectos de nuevos medicamentos, nuevas alternativas quirúrgicas, suplementación nutricia, entre otros tipos de intervención. Asimismo, por el tipo de aleatorización, los ECCA pueden ser aleatorizados y no aleatorizados. Existen al menos tres tipos de asignación de la intervención: fija, dinámica y adaptativa. Por el tamaño de muestra, los ECCA pueden clasificarse en fijos y secuenciales; finalmente, por el número de sedes, pueden ser de sitio único y multicéntricos (cuadro I). A continuación describiremos los ECCA por estructura de tratamiento.

Ensayo clínico controlado aleatorizado, por estructura de tratamiento
Con diseño paralelo
En los ECCA de tipo paralelo, los sujetos de estudio siguen el tratamiento al que han sido asignados al azar durante el tiempo que dure el ensayo.
Con diseño de tratamiento sucesivo
En los ECCA de tratamiento sucesivo cada sujeto es asignado al azar a un grupo que sigue una secuencia de tratamiento previamente determinada, de manera que cada persona recibe más de un tratamiento. La forma más frecuente es el diseño de tratamiento sucesivo en dos periodos, con un primer tratamiento seguido de un segundo. Entre el primero y el segundo se deja un periodo sin tratamiento, de forma que se disipen los efectos residuales del primero. A este respecto, existen básicamente dos tipos de ECCA de diseño de tratamiento sucesivo: el diseño de tratamiento de remplazo y el diseño cruzado.
El diseño de tratamiento de remplazo se usa para recolectar datos sobre efectos que tiene el cambiar de un tratamiento A sobre uno de dos tratamientos alternativos, por ejemplo, tratamiento B o tratamiento C. Los sujetos de estudio se dividen en dos grupos iguales. Ambos grupos reciben el tratamiento A durante un primer periodo. Las observaciones hechas entre los pacientes tratados con A y B se comparan con los resultados observados entre los pacientes tratados con A y C (figura 2).

En ECCA con diseño cruzado, el grupo 1 recibe el tratamiento A durante un primer periodo y el tratamiento B en el segundo. El grupo 2 recibe los tratamientos en orden inverso al grupo 1. Los diseños cruzados permiten ajustar las variaciones de persona a persona haciendo que cada sujeto sirva como su propio control. En este diseño se exige con frecuencia un menor número de sujetos en relación con otros diseños, y su esencia es que cada sujeto sirve como su propio control (figura 3). Un ejemplo de esta estrategia es el estudio realizado en México para evaluar el impacto nutricio de una intervención comunitaria sobre el crecimiento en niños de bajo ingreso económico menores de 12 meses de edad en seis estados del centro del país.5 Mediante un proceso de aleatorización 205 comunidades fueron asignadas para el grupo de intervención y 142 comunidades lo fueron para el grupo de intervención cruzada. El grupo de intervención recibió el programa durante dos años (primer periodo, primer año), mientras que el grupo de intervención cruzada solamente lo recibió durante el segundo año (segundo periodo). Los resultados mostraron un crecimiento mayor en el grupo de intervención (26.4 cm) que en el grupo de intervención cruzada (25.3 cm), así como valores medios de hemoglobina mayores (11.12 vs. 10.9, respectivamente).

Diseños alternativos
Diseño factorial
La evaluación de dos o más intervenciones en el mismo ECCA puede ser implementada usando un diseño de tipo paralelo. Sin embargo, se requiere aumentar el tamaño de muestra y puede ser ineficiente, especialmente si hay también interés en considerar combinaciones de las intervenciones. El diseño alternativo en esta situación es de tipo factorial, donde se pueden asignar de manera aleatoria dos o más intervenciones en forma independiente, siempre y cuando no exista una interacción; de tal manera que los sujetos pueden no recibir ninguna intervención, una de ellas o eventualmente todas.6 En la figura 4 se refiere un ejemplo de un ensayo factorial, donde cada paciente es aleatorizado dos veces para recibir dos tratamientos en el mismo ensayo;* esto es, bajo el supuesto de que no hay interacción, dos experimentos pueden ser conducidos en uno. Se trata de la evaluación de una intervención para prevenir paludismo y anemia, con quimioprofilaxis de deltaprim y hierro, respectivamente. Los autores establecieron con estos resultados que la quimioprofilaxis de malaria durante el primer año de vida es efectiva en la prevención de paludismo y anemia, y que la suplementación con hierro es efectiva para prevenir anemia severa sin incrementar la susceptibilidad a malaria.7

Diseño de equivalencia
Se ponen en práctica para demostrar que dos tratamientos son efectivamente similares respecto a la respuesta del paciente. Son diseños no sesgados que evalúan diferencias en tratamiento cercanas a cero y con un estrecho intervalo de confianza. Se ponen en práctica porque existen tratamientos que pueden diferir en seguridad, efectos adversos, conveniencia de administración, costos, entre otras características; y el hecho de mostrar "equivalencia" tiene importancia para el uso subsiguiente de uno o ambos tratamientos. Un ejemplo de ello es el ensayo clínico realizado en Japón para probar la eficacia del maleato de timolol con sorbato de potasio (MTSP), comparado con maleato de timolol (MT) en sujetos con hipertensión ocular, incluyendo aquellos con glaucoma de ángulo abierto, con edad igual o mayor a 18 años, con afectación ocular uni o bilateral (presión ocular igual o mayor a 22 mm de Hg). A un grupo se le administró a nivel ocular MT una vez al día y el otro grupo MTSP dos veces al día durante 12 meses. Al final del periodo del estudio, 95% de los pacientes recibieron los medicamentos asignados; los resultados entre ambos grupos fueron similares, y se encontró una reducción de la presión ocular en ambos brazos del estudio.8
Fases de un ensayo clínico para evaluar efectos terapéuticos de nuevos fármacos
La investigación clínica de evaluación de un nuevo agente terapéutico (incluidas vacunas), previamente no evaluado, es generalmente dividida en cuatro fases (cuadro II). Aunque las fases pueden ser conducidas secuencialmente, en algunas situaciones se pueden traslapar.

La fase I incluye el inicio de estudio de un nuevo agente farmacológico en un grupo de entre 20 y 80 sujetos. Son cercanamente monitoreados y pueden ser conducidos en sujetos sanos o con condiciones mórbidas. Esta fase del estudio es diseñada para determinar las acciones farmacológicas, el metabolismo de las drogas en humanos, así como los mecanismos de acción, las reacciones adversas asociadas con el incremento de dosis y, si es posible, obtener evidencia temprana de su efectividad. Asimismo, también incluye estudios en los que los nuevos agentes farmacológicos son utilizados como herramientas de investigación para explorar fenómenos biológicos o el proceso de enfermedad. Durante esta fase se cuantifican ampliamente los efectos farmacocinéticos y farmacológicos que permitirán planear la fase subsiguiente.
La fase II incluye los estudios clínicos controlados, conducidos para evaluar la efectividad de las drogas para una particular indicación en pacientes con la enfermedad o condición bajo estudio; para determinar los efectos adversos más comunes y los riesgos asociados con el uso de estos nuevos agentes farmacológicos. Esta fase debe ser bien controlada, cercanamente monitoreada y conducida en un pequeño número de sujetos. Puede subdividirse en fase IIA, donde se decide si el tratamiento u otro procedimiento en particular son suficientemente efectivos para justificar un estudio adicional. Para ello se fija un nivel de efectividad, y a partir de éste se evalúa la posibilidad de encontrar 95% de éxitos o, por el contrario, se admite 5% de fracasos. La fase IIB es desarrollada para estimar la efectividad y la magnitud de la misma. Con esta información es posible planear tamaños de muestra en estudios de fase III.
La fase III es realizada cuando existe evidencia preliminar que sugiere efectividad del nuevo agente farmacológico obtenido, y se pretende ganar información adicional acerca de la seguridad y efectividad que son necesarias para evaluar la relación beneficio-riesgo. Esta fase de estudio es desarrollada generalmente con un gran número de sujetos.
Los estudios clínicos fase IV incluyen todas las investigaciones realizadas después de la aprobación del medicamento; en otras palabras, son los estudios de medicamentos de uso rutinario o también se conocen como estudios de posmercadeo.9,10 El objetivo de estos estudios está muy definido, obtener conocimiento adicional de la eficacia y seguridad de un medicamento.9 La información obtenida acerca de un medicamento en los estudios fase I a III no proporciona bases suficientes para establecer conclusiones finales acerca del valor clínico de un medicamento posterior a su comercialización. En comparación con la fase III, la cual tiene un tipo de diseño clásico de ECCA, la fase IV requiere de diferentes diseños: reportes de casos, series de casos, estudios de observación comprensiva, estudios de casos y controles, estudios de cohorte, análisis de perfil de prescripción y de reporte de eventos adversos, análisis comparativo de bases de datos y estudios de costo beneficio. Estos son algunos ejemplos de tipos de diseño utilizados en esta fase.10
Características metodológicas
Al definir la hipótesis primaria de un ECCA se ha sugerido que los investigadores no sólo establezcan una hipótesis nula de no efectos en el tratamiento en la comparación de grupos, sino que con base en una revisión sistemática puedan elegir algunas hipótesis secundarias alternativas, claramente definidas antes de iniciar el estudio.
Asimismo, para evaluar si el diseño es apropiado para responder a la pregunta de investigación deben considerarse los siguientes aspectos: a) definición del evento resultado primario, b) disponibilidad del protocolo de tratamiento bajo estudio, y c) identificación de la población elegible. Un claro ejemplo de este hecho es que si deseamos implementar una intervención para evitar la progresión de una enfermedad, debiéramos reclutar sujetos con evidencia de encontrarse en fases tempranas de la enfermedad bajo estudio y no incluir a aquellos que están en riesgo de sufrirla.
Por estas razones se han implementado diversas estrategias para evaluar la pertinencia en el planteamiento y reporte de ensayos clínicos aleatorizados. La publicación de un ECCA debe transmitir al lector, de manera clara, por qué el estudio fue llevado a cabo y cómo fue conducido y analizado.11 Un grupo de investigadores clínicos propusieron, en 1981, guías clínicas para usuarios de la literatura médica, con el fin de evaluar críticamente la información de artículos sobre diferentes tópicos incluidos estudios sobre tratamiento.12 Por otro lado, a mediados de 1990 dos iniciativas independientes para mejorar la calidad de las publicaciones de ECCA, el grupo para establecer las Normas para Información sobre Ensayos (SORT, por sus siglas en inglés) y el grupo de trabajo Asilomar, encargado de las recomendaciones para los Informes de los Ensayos Clínicos en la Literatura Biomédica, desarrollaron en forma conjunta las Normas Consolidadas para la Publicación de Ensayos clínicos, las cuales fueron publicadas en 1996.13 La Declaración CONSORT comprende una lista de comprobación de 22 puntos y un diagrama de flujo para comunicar un ECCA. Por conveniencia, la lista de comprobación y el diagrama juntos son denominados sencillamente CONSORT y principalmente han sido diseñados para escribir, revisar y evaluar informes de ECCA simples de sólo dos grupos paralelos. La lista de comprobación y el diagrama de flujo pueden ser observadas en el cuadro III y la figura 5.

Cegamiento
El cegamiento es una condición impuesta sobre un procedimiento específico para intentar guardar el conocimiento del tratamiento asignado, el curso del tratamiento u observaciones previas.14 Los procedimientos usualmente cegados son la intervención asignada o evaluación del estatus de los sujetos de estudio. El cegamiento previene determinados sesgos en las diversas etapas del ensayo clínico y protege la secuencia después de la asignación al grupo de tratamiento. A este respecto, existen básicamente tres niveles de cegamiento, entre los cuales se encuentran el simple, el doble y el triple, cuyas características son descritas en el cuadro IV.

Placebo
Un placebo es un agente farmacológicamente inactivo que los investigadores administran a los participantes en el grupo control de un ensayo clínico. El primer ensayo clínico controlado con placebo fue conducido probablemente en 1931, cuando se probó el sanocrysin en comparación con agua destilada en pacientes con tuberculosis.15 Desde entonces, los ensayos clínicos aleatorizados con placebo han sido controversiales, especialmente cuando los participantes son asignados de manera aleatoria en una de sus ramas [placebo por ejemplo] y por lo tanto se les priva del tratamiento efectivo.16,17 En los ensayos clínicos el placebo se traduce en tratamientos de control con similar apariencia a los tratamientos en estudio, pero sin su actividad específica.18 El uso de placebo ha reportado resultados objetivos y subjetivos en un rango de 30 a 40% de los pacientes en una amplia gama de entidades clínicas, entre las que destacan dolor, asma, hipertensión arterial sistémica e, incluso, infarto al miocardio, entre otros.19 Desde hace 50 años se ha documentado que el uso de placebo tiene un alto grado de efectividad, interpretado bajo mecanismos desconocidos como un efecto terapéutico real en más de 30% de los casos.20 Por esta razón, el efecto placebo no podrá ser distinguido de la historia natural de la enfermedad, regresión a la media o el efecto de otros factores. El debate acerca del uso apropiado del placebo en las investigaciones clínicas aparece posterior a la citación de numerosos ejemplos de ensayos clínicos en los cuales se empleaba placebo a pesar de la existencia de un tratamiento efectivo.21 Dichos estudios violaban principios básicos de la Declaración de Helsinki. Además, la aparición del VIH-SIDA y las innovadoras metodologías para evaluar drogas de reciente aparición (ensayos clínicos multinacionales, financiadores externos al país huésped, uso de comparadores-placebo, por ejemplo), sobre todo en las naciones con menores ingresos, provocó debates éticos por parte de los miembros de las instancias reguladoras internacionales (cuadro V).

Aleatorización
El propósito primario de la aleatorización es garantizar que la posible inferencia causal observada al final del estudio no se deba a otros factores. Una gran variedad de procedimientos de aleatorización se han sugerido en la literatura.
La aleatorización se refiere a la asignación a través del azar de las unidades de investigación a uno de dos o más tratamientos, con la finalidad de comparar los tratamientos sobre las variables de desenlace de interés. Se acepta que la aleatorización tiene como propósito prevenir la existencia de diferencias entre los grupos que no sean derivadas de los tratamientos que se están comparando. De esta manera, cuando se produce un equilibrio de las posibles variables que pudieran modificar el efecto del tratamiento sobre la variable de desenlace, las diferencias que se encuentran se deben considerar estrictamente como debidas a la maniobra bajo estudio.
El concepto de aleatorización fue originalmente utilizado por Fisher en su texto clásico El diseño de experimentos, teniendo como argumento principal que la aleatorización prevendría las diferencias sistemáticas de cualquier tipo, independientemente de que éstas pudieran ser identificadas por el investigador. Este concepto convierte al proceso de aleatorización en preferible sobre la asignación no probabilística (sistemática, secuencial, por facilidad o por conveniencia), la cual en ningún momento tiende a asegurar equilibrio entre los grupos.
Sin embargo, siempre hay que tener cuidado, ya que la aleatorización puede no resultar en una distribución equilibrada de las variables confusoras entre los grupos, por lo que siempre es obligado efectuar una comparación de las variables al ingreso del paciente al estudio que pudieran afectar los efectos de la maniobra y, en caso de que existan diferencias significativas, ajustar los resultados obtenidos por dichas variables. A continuación se describen los métodos utilizados para asignar la maniobra de intervención.24-35
Asignación aleatoria de los tratamientos
Un objetivo importante de la investigación clínica es el desarrollo de terapéuticas que mejoren la probabilidad de desenlaces exitosos en los sujetos enfermos o que prevengan el inicio de la enfermedad en los individuos sanos. La evidencia convincente de la efectividad de una maniobra requiere no sólo observar una diferencia entre los grupos respecto al desenlace de interés, sino también demostrar que la maniobra es la que más probablemente ha causado dichas diferencias. Por ejemplo, los pacientes que acaban de ser sometidos a una nueva intervención quirúrgica pueden presentar un mayor tiempo de supervivencia que los que recibieron la intervención convencional. Habría que analizar hasta qué grado el resultado de la supervivencia depende de la cirugía y no de la habilidad del cirujano para seleccionar a los pacientes de bajo riesgo quirúrgico.
Para asegurar una evaluación no sesgada de los tratamientos, los grupos de estudio deben ser equivalentes en todo, excepto en las maniobras que están recibiendo (ceteris paribus).
Necesidad de aleatorización en los ensayos clínicos
En muchos experimentos realizados en el laboratorio, los científicos poseen las herramientas para lograr equivalencia entre las unidades bajo investigación. Con la capacidad para mantener un control perfecto sobre las muestras que están siendo comparadas, experimentos pequeños ejecutados fastidiosamente pueden ser suficientes para medir en forma precisa los efectos de las maniobras. En biología, sin embargo, especialmente cuando los sujetos bajo estudio son los seres humanos en su totalidad, la variabilidad inherente ocasiona que el control de los potenciales confusores sea prácticamente imposible. Si bien existen algunas estrategias para parear grupos asignados a las diferentes maniobras de acuerdo con los posibles confusores, esto generalmente requiere tamaños de la muestra elevados en cada estrato y, aun así, debido a la existencia de variables desconocidas, según el estado actual del conocimiento, hace al balance imposible.
En adición a la dificultad de tratar de seleccionar grupos comparables siempre se debe cuidar la preferencia consciente o inconsciente que puede tener el médico por pacientes específicos de acuerdo con el tratamiento por probar, lo cual puede sesgar el resultado de la investigación. Por lo anterior, una característica importante de la asignación de los pacientes a los tratamientos debe ser la falta de predictibilidad, es decir, que la asignación del siguiente tratamiento siempre debe decidirse a través de los procesos de aleatorización.
Aleatorización como base para la inferencia estadística
De acuerdo con la teoría frecuentista, la aleatorización permite realizar pruebas directas de causa y efecto y construir pruebas válidas de significancia estadística. Por ejemplo, en un ensayo que evalúa el efecto de un nuevo medicamento sobre el nivel de LDL-colesterol, el modelo inicia efectuando una medición de los niveles basales de LDL-colesterol en cada paciente. A través de un proceso de aleatorización cada paciente es asignado a recibir el nuevo medicamento de interés o la maniobra comparativa, y al final del periodo de estudio se vuelven a medir los niveles de LDL-colesterol en cada paciente. Si lo que se esperaba era que el medicamento redujera LDL-colesterol en 10 mg/dl, entonces el grupo al que se le administró el nuevo medicamento deberá tener en promedio niveles de LDL-colesterol menores que el grupo control. Para probar este efecto uno compara los promedios observados en los dos grupos. Gracias al proceso de aleatorización, que en términos generales balancea entre los grupos los posibles confusores, uno espera que si las diferencias son suficientemente grandes y significativas se pueda concluir que el tratamiento las ha producido.
Métodos de aleatorización
La aleatorización requiere un mecanismo gobernado por el azar para asignar las maniobras (los tratamientos) a los sujetos bajo investigación. Los ensayos clínicos reales deben utilizar métodos verificables de aleatorización, de tal manera que después del estudio el investigador pueda demostrar que la asignación se mantuvo libre de sesgo.
Asignación aleatoria simple
La manera más sencilla de asignar la maniobra de intervención es la aleatorización simple. En ella se utiliza como herramienta base la tabla de números aleatorios. Se selecciona al azar un punto de inicio y posteriormente se selecciona la dirección de movimiento que se mantendrá constante a lo largo de toda la tabla. En forma a priori se decide qué grupos de números (0 al 9) se destinarán a cada maniobra, pudiendo quedar por convenido que los números pares (0, 2, 4, 6 y 8) se destinarán a la maniobra A y los nones (1, 3, 5, 7 y 9) a la maniobra B. Así, por ejemplo, en el cuadro VI se incluye una sección de una serie de números aleatorios contenida en cualquier libro de bioestadística básica, seleccionándose por azar el punto de inicio en el segundo renglón, de la segunda fila y en el primer número 5 que aparece. En este caso se selecciona en dirección de izquierda a derecha y arriba hacia abajo de modo que partiendo del 5, los números que continúan son 6, 8, 9, 7 y 2. De esta manera, dado que el primer número a emplear es el 5, el primer sujeto que ingrese al estudio recibirá la maniobra B, el segundo la maniobra A, el tercero la maniobra A, el cuarto la maniobra B y así sucesivamente hasta asignar al total de sujetos necesarios a incluir.

Debido a lo tedioso que puede resultar efectuar la asignación manual, cuando se trata de una gran cantidad de sujetos y que de acuerdo con algunos investigadores puede ser sujeta de algunos tipos de sesgo, cuando sea posible, una computadora debe producir la lista de aleatorización. La persona que trabaja en el equipo de cómputo para generar la lista de aleatorización debe ser ajena a las personas que reclutan y valoran a los participantes en la investigación. Durante el curso del estudio el generador de las listas no debe divulgar los detalles del método particular utilizado para generarlas.
Muchos estudios utilizan sobres opacos en cuyo interior se encuentra cuál será el siguiente tratamiento por asignar. Este método, aunque considerado por muchos investigadores como estándar, puede ser sujeto a violación, especialmente en estudios no cegados. Un investigador que desea que determinado tipo de pacientes ingresen a una rama específica del estudio puede observar a contraluz el contenido del sobre o incluso abrirlo y volverlo a cerrar. Lo anterior ha generado que muchos estudios en la actualidad adopten códigos de aleatorización generados vía telefónica, por fax o encriptados a través de sistemas de cómputo.
Una de las desventajas de este tipo de aleatorización es que cuando las muestras son pequeñas por lo general se producen desbalances en el número de sujetos asignados a cada tratamiento, pudiendo asignarse un mayor número de sujetos a determinada maniobra. Otra limitante importante es que en ocasiones se producen secuencias repetidas de una misma maniobra (sujetos 4, 5 y 6 a maniobra A, luego sujetos 7 y 8 a maniobras B, luego sujetos 9, 10, 11,12 y 13 a maniobra A, y así sucesivamente). Los sujetos que ingresan en un determinado tiempo al estudio pueden ser distintos en sus características basales o por la forma de responder a las maniobras, en relación con los sujetos que ingresan a la maniobra alternativa.
Aleatorización en bloques balanceados
Tratando de limitar la posibilidad de desbalances en la asignación de tratamientos, de generar secuencias repetidas largas de una misma maniobra y de balancear en la medida de lo posible algunos de los sesgos inherentes al proceso de aleatorización simple se creó el método de aleatorización en bloques balanceados. En este método se ensambla una serie de bloques, formados por un número determinado de celdas, en las cuales se incluyen los distintos tipos de tratamiento. El número de bloques estará determinado por el número de participantes a incluir en el estudio y el número de celdas que se haya decidido incluir en cada bloque. Cada bloque contendrá en cada celda una de las alternativas de tratamiento y dentro de cada bloque deberá existir un número balanceado de los posibles tratamientos (cuando por fines éticos y de seguridad se considera conveniente asignar el doble o triple de pacientes a una determinada maniobra, se dice que se trata de una aleatorización en bloques desbalanceados, ya que existirá el doble o triple de celdas en cada bloque de uno de los tratamientos).
Así por ejemplo, en el cuadro VII se presenta en caso de aleatorización en bloques balanceados donde se pretenden asignar 24 sujetos a dos alternativas de tratamiento, decidiéndose utilizar bloques balanceados con longitud fija de cuatro celdas por bloque. Puesto que se trata de 24 pacientes y se incluirán cuatro celdas en cada bloque se necesitarán seis bloques (número de bloques= número de pacientes/número de celdas por bloque). Dado que se incluirán cuatro celdas por bloque y sólo existen dos alternativas de tratamiento se deberá incluir en cada bloque las diferentes combinaciones de A y B (ver ejemplo). Quien asigna el número de uso a cada bloque es la tabla de números aleatorios, así que por azar el primer número del tercer renglón de la primera columna es el número 2, por lo tanto el primer bloque es el número 2, el siguiente bloque es el 4, el tercer bloque es el 6 en uso y luego, debido a que los números siguientes son el 7 y 8, éstos no se utilizan, sigue el número 1 (para el cuarto bloque, luego el 0 no se usa y si se decide continuar en la dirección marcada con la flecha los números que siguen es el 1 (repetido) y luego el 3 (para el quinto bloque) y, finalmente, el sexto bloque es el 5. Una vez asignado el número a cada bloque se utilizan las combinaciones de tratamientos contenidas dentro de ellos.

Aleatorización estratificada
Si bien la aleatorización en bloques balanceados permite la asignación balanceada de los sujetos a las maniobras, independientemente del momento en el que se decida detener el ensayo y de cuántos pacientes se hayan incluido hasta ese momento, sigue manteniendo la desventaja de que no permite efectuar un balanceo por las posibles variables modificadores de efecto o confusoras. Esto ha llevado a que diversos investigadores señalen que es mejor identificar a través de la revisión sistemática o narrativa de la literatura, de la consulta con expertos o de la experiencia misma, aquellos factores que en un momento dado pudieran modificar el impacto de la maniobra sobre la variable de desenlace y, dependiendo de la factibilidad del tamaño de la muestra, decidir cuántos estratos establecer en forma a priori a la asignación de la maniobra (y en cuantos se efectuará ajuste de los resultados al momento del análisis si es que posterior a la asignación aleatoria quedan desbalanceados). Así, por ejemplo, el cuadro VIII contiene un ejercicio en el cual se ha identificado la edad mayor de 60 años, el género masculino, la historia familiar y la presencia de dificultad respiratoria al momento de ingreso al estudio como potenciales modificadores de los efectos de la maniobra sobre la variable de desenlace. Se establecen los diferentes subestratos y al final de la estratificación se efectúa la asignación de la maniobra a través de asignación simple o en bloques balanceados (dependiendo del número de pacientes disponibles y de la posibilidad de desbalances en la asignación), pudiéndose observar cómo inmediatamente este método degenera, conforme el número de estratos aumenta (por ejemplo, cuando se tienen cinco posibles confusores con tres categorías para cada uno, se tendrá la necesidad de ensamblar 35=243 distintos estratos).
Aleatorización en conglomerados (grupos o clusters)
Se trata de un proceso de aleatorización simple o en bloque de grupos de personas, salones, delegaciones, comunidades, municipios, ciudades, estados o países. La unidad de asignación es el grupo y no el individuo. Este tipo de aleatorización es ampliamente utilizada en investigaciones epidemiológicas, también puede emplearse para evaluar el impacto de programas educativos, medidas preventivas comunitarias, entre otras.
No obstante que para fines de aleatorización la unidad de interés es el grupo y no los individuos contenidos dentro de ellos, es importante medir el grado de similitud de respuesta dentro del conglomerado. Para ello se utiliza el cálculo del coeficiente de correlación intraclase o intraconglomerado denotado por la letra griega r. Este parámetro puede ser interpretado como el coeficiente de correlación estándar de Pearson entre cualesquiera de dos respuestas en el mismo conglomerado. En términos generales, cuando el valor de r es positivo se asume que la variación entre las observaciones en diferentes conglomerados excede la variación dentro de los conglomerados. Las razones para ello pueden deberse a la manera de seleccionar los sujetos, la influencia de covariados en el conglomerado, la posibilidad de compartir algunos factores más rápidamente dentro que entre las comunidades o al efecto de las interacciones personales entre los miembros del conglomerado que reciben la misma maniobra.
Asignaciones dinámicas o adaptativas de los tratamientos
Los diseños más simples de los ensayos clínicos controlados consideran la inclusión de un predeterminado número de individuos bajo estudio, los cuales son asignados en alguna de las formas tradicionales ya descritas a alguno de los tratamientos de interés con igual probabilidad para cada paciente de recibir una u otra modalidad terapéutica. En forma cada vez más frecuente, tanto la manera como los sujetos de estudio son asignados a los tratamientos, como la forma en la que se decide terminar el estudio se basan en la información que se va generando y acumulando durante el progreso del estudio. Así, se habla de asignación dinámica cuando la información sobre los covariados del paciente que predicen el desenlace clínico se utiliza para determinar la asignación del tratamiento. Por el contrario, se habla de asignación adaptativa cuando se utilizan datos de desenlace acumulados que afectan la selección del tratamiento.
La asignación dinámica permite efectuar un balance individual de los posibles confusores sin tener que efectuar un balance dentro de todas las combinaciones de los factores (como en el caso de la aleatorización estratificada tradicional). Supóngase que existen f factores y lf niveles en el factor f. A cualquier punto dado en el ensayo, la asignación del tratamiento del paciente previo habrá creado algo de desbalance entre los factores. Si dejamos que sea tijk el número total de pacientes en el jth nivel del factor i que ha sido asignado al tratamiento k, i=1, ……….,f, j=1,……,lf, k=1,…..,r, donde r es el número de tratamiento, el ensayo está balanceado para el factor i nivel j al grado de que tij1,…..,tijr son similares. Si el siguiente paciente a ser aleatorizado posee el factor i al nivel jth, entonces puede considerarse el efecto que cada posible asignación de tratamiento tendría en este balance. Dado que el balance debe ser caracterizado por una función matemática, se ha propuesto un método de minimización, donde el balance es caracterizado por un rango de tratamientos totales y el tratamiento es seleccionado por minimización de la suma a través de todos los factores. Existe también una versión más general de este método en el cual el tratamiento se selecciona a través de una aleatorización por moneda sesgada, con las probabilidades de la moneda sesgada determinadas por la función de balanceo. La función de balanceo general involucra una suma ponderada de las funciones de balanceo de los factores individuales, donde los pesos podrían ser asignados sobre la base de la importancia relativa de cada factor confusor.
Por su parte, los diseños adaptativos, los cuales dependen de la acumulación de datos de desenlace, han sido descritos desde principios de 1950. Así, Armitage propuso un esquema que permitía la terminación global del estudio basado en un nivel de significancia global, dando origen a los famosos análisis intermedios (análisis interim) en los cuales se producía una culminación prematura del estudio cuando se alcanzaban ciertos límites de desenlace y significancía estocástica. En 1963 estas ideas fueron revolucionadas por Colton quien propuso reglas de detención de los ensayos basadas en pérdidas de la función adecuada, en contraste con las reglas estocásticas utilizadas previamente (de significancia estadística). Para ello se propuso la construcción de un "horizonte del paciente", en el cual los pacientes continuaban siendo aleatorizados hasta que se cruzaban los límites pre-establecidos, después de lo cual todos los pacientes restantes se asignaban al tratamiento con la mayor eficacia. Así, los límites óptimos se establecían al intercambiar las pérdidas generadas por aleatorizar la mitad de los pacientes al tratamiento inferior. En 1969 Zelen popularizó este método de aleatorización bajo el nombre de sistema "Jugando al ganador", en el cual el primer paciente se asignaba por ejemplo a la maniobra A, si se obtenía un éxito, el siguiente paciente se continuaba asignando al grupo A, hasta que se tuviera una falla en la cual el siguiente paciente se asignaba a B y así sucesivamente.
Análisis de un ensayo clínico
"Un ensayo clínico apropiadamente planeado y ejecutado es una técnica experimental poderosa para estimar la efectividad de una intervención".36 Este concepto ha sido aplicado a numerosos estudios realizados en todo el mundo, bajo la premisa de que todo ensayo clínico controlado comienza con la planeación cuidadosa del mismo, pasando por un proceso detallado de ejecución y monitoreo, sin menospreciar cualquier procedimiento por simple que parezca para garantizar la comparabilidad de los datos obtenidos. La estimación de los resultados se realiza a través de diferentes técnicas estadísticas que comentaremos en el presente capítulo, sin embargo, es imprescindible mencionar que la piedra angular de un análisis estadístico axiomático está fundamentada en el meticuloso planteamiento del diseño.
Interpretación de ensayos clínicos
Principio analítico por intención de tratar
Los ensayos clínicos aleatorizados son analizados por un método estándar llamado "intención de tratar", esto es, todos los sujetos aleatorizados son analizados de acuerdo con la asignación original del tratamiento y todos los eventos son contados contra el tratamiento asignado.37 Con base en este principio, todos los estudios aleatorizados deberían ser analizados bajo este concepto, ya que el análisis apoyado por la aleatorización mantiene la comparabilidad a través de los grupos de intervención. Si el análisis excluye a participantes después del procedimiento de aleatorización, (por razones tales como sujetos que no reciben el tratamiento originalmente asignado o el participante muere antes de que el tratamiento sea dado) se puede introducir un sesgo, ya que los grupos de intervención se verán afectados por la falla en el cumplimiento del diseño que tomó originalmente en consideración la aleatorización, el tamaño de la muestra y el blindaje de los grupos.38
Entre las razones más comunes para que pacientes aleatorizados sean excluidos está el que no se consideren elegibles, posterior al procedimiento aleatorio. Citemos el trabajo del Anturane Reinfarction Trial Research Group39,40 el cual evaluó el efecto de la sulfinpirazona en pacientes que habían sufrido un infarto. La aleatorización incluyó en dos grupos a 1 629 pacientes quienes sobrevivieron a un infarto del miocardio, uno manejado con el tratamiento de prueba y otro con placebo. Los pacientes clasificados como elegibles fueron 1 558, mientras que 71 no reunieron los criterios de elegibilidad del protocolo. El análisis reportado se enfocó solamente a los pacientes elegibles y mostró un efecto benéfico de la sulfinpirazona sobre la mortalidad, comparado con los no elegibles; sin embargo, un análisis posterior realizado por otros investigadores41 reportó que la inclusión de aquellos no elegibles modificaba completamente los resultados, mostrando que la interpretación de los resultados estaba sesgada por la exclusión, posterior a la aleatorización, de los sujetos declarados como no elegibles. Los resultados de este estudio fueron cuestionados por una agencia de regulación federal.
Otra de las razones para la exclusión de pacientes en el análisis primario aparece cuando los pacientes no cumplen apropiadamente con la intervención especificada en el protocolo (falta de apego al tratamiento). Citemos el trabajo del Coronary Drug Project,42 cuyo objetivo fue evaluar una estrategia medicamentosa para reducir el colesterol en hombres que sobrevivieron a un infarto del miocardio. El estudio tuvo dos grupos, uno tratado con bezofibrato y otro con placebo. Los resultados globales no mostraron diferencias en la mortalidad. Sin embargo, un análisis extra clasificó en dos grupos a los pacientes: uno de ellos como buenos cumplidores del tratamiento (definido como que tomó en más de 80% sus medicamentos) y otro grupo como poco cumplidor (<80%). La comparación de los poco cumplidores para tomar bezofibrato mostró una reducción de la mortalidad de 24.6 a 15%, sin embargo, una reducción mayor en la mortalidad fue observada en el grupo placebo (28.2% vs 15.1%). Los buenos cumplidores vivieron más que los poco cumplidores, independientemente del tratamiento. Estos resultados no pudieron ser explicados por modelos de regresión y los autores concluyeron que el apego a una intervención es, en sí misma, un resultado.
Aun cuando las exclusiones sean previstas a priori en el protocolo, deberá tomarse en cuenta un posible sesgo, ya que los sujetos excluidos pueden diferir de los sujetos analizados en aquellas características medidas y no medidas, las cuales no siempre se pueden probar estadísticamente debido a que el no apego al tratamiento no es un fenómeno aleatorio. Ningún método de análisis puede completamente contabilizar el gran número de sujetos de estudio que se desviaron del protocolo de estudio, lo que resulta en altas tasas de no apego, abandono o datos faltantes.
Recomendaciones para ajustar el diseño y aumentar el tamaño de la muestra pueden compensar el no apego. Se debe contemplar en el diseño elegir un método de intervención con mejor tolerancia, así como plantear estrategias de monitoreo durante la toma del tratamiento. Otra estrategia es aumentar el tamaño de la muestra para compensar el efecto del no cumplimiento de los tratamientos. Se requiere un incremento de 23% en el tamaño de la muestra para compensar 10% de no apego, mientras que 20% de no apego requerirá un incremento del tamaño de muestra en 56%.43
El principio de análisis por intención de tratar es entonces el método de análisis más conservador y provee una estimación de la efectividad del tratamiento (efecto del tratamiento dado a cada participante) más que una prueba verdadera de la eficacia del mismo (esto es, efecto del tratamiento en aquellos quienes siguieron el protocolo de estudio).
Pérdidas en el seguimiento
Aunque apropiadamente planeado para tener un mayor tamaño de muestra que compense pérdidas en el seguimiento, cuando éstas ocurren, el estudio puede sufrir pérdida de poder estadístico y aumentar la posibilidad de introducir un sesgo, primordialmente cuando la pérdida no es resultado del azar. Los pacientes más enfermos pueden no estar disponibles para regresar a la clínica y realizarse exámenes y mediciones clave para el resultado del estudio. En otros casos, pacientes sometidos a toxicidad medicamentosa pueden estar indispuestos para ser evaluados en las mediciones clave. Cuando éstos son los casos, los resultados pueden estar sesgados y ningún método estadístico podrá ajustar por los datos faltantes. Entonces, se requerirán esfuerzos extras para localizar y estimar la variable de resultado primaria en aquellos participantes que no recibieron la intervención.
Análisis por protocolo
El análisis por protocolo es un método de análisis que incluye solamente a un subgrupo de sujetos quienes cumplieron suficientemente con el protocolo por lo que contrasta con el análisis conservador por intención de tratar. El cumplimiento del protocolo incluye el cumplimiento de exposición mínima pre-especificada al régimen de tratamiento, disponibilidad de mediciones de la variable primaria, elegibilidad correcta y ausencia de cualquier otra violación mayor al protocolo.44 Excluye también eventos que ocurrieron después de que el sujeto dejó de apegarse al protocolo. También puede ser identificado este método como de "casos válidos", muestra de "eficacia" o de "sujetos evaluables".
Este método puede maximizar la oportunidad para un nuevo tratamiento que muestre eficacia adicional y refleje más cercanamente el modelo científico fundamental. Sin embargo, la correspondiente prueba de hipótesis y estimación del efecto del tratamiento puede o no ser conservadora dependiendo del ensayo; el sesgo, el cual puede ser severo, emana del hecho de que el apego al protocolo puede estar relacionado con el tratamiento y por ende al resultado.
Las razones precisas para la exclusión de los sujetos del grupo por protocolo deben ser cuidadosamente definidas y documentadas antes de romper el blindaje en una forma apropiada dentro de las circunstancias del ensayo específico. Los problemas que guían a la exclusión de los sujetos para crear el subgrupo por protocolo, y otras violaciones al protocolo de estudio, deben ser cuidadosamente identificados y analizados. Violaciones relevantes al protocolo pueden incluir errores en la asignación del tratamiento, uso de medicamentos excluidos, pobre cumplimiento, pérdidas en el seguimiento y datos faltantes. Es una buena práctica estimar el patrón de tales problemas entre los grupos de tratamiento con respecto a la frecuencia y tiempo de ocurrencia.
Resultados de variables subrogadas
Debido a que muchos ensayos clínicos usan como resultados variables clínicas que son evaluadas por largo tiempo y aumentan los costos, algunos investigadores han visto alternativas para obtener ensayos clínicos más cortos y más pequeños a través del uso de una variable respuesta sustituta del resultado clínico. Un requerimiento para obtener esta variable sustituta es que debe ser predictiva del resultado clínico. Otro requerimiento es que la variable sustituta debe capturar el efecto total de la intervención sobre el resultado clínicamente relevante. Aunque teóricamente posible, biológicamente es un fenómeno complejo donde la intervención puede modificar la variable subrogada y no tener efecto o tenerlo parcialmente sobre la variable clínica de respuesta. Por el contrario, la intervención puede modificar la variable clínica sin afectar a la subrogada.
Aunque se han propuesto criterios estadísticos para validar las variables subrogadas, la experiencia con su uso es limitado. En la práctica, la fuerza de la evidencia para la sustitución depende de tres aspectos: a) la plausibilidad biológica de la relación entre la variable clínica/variable subrogada; b) la demostración en estudios epidemiológicos del valor pronóstico para la variable clínica, y c) evidencia de ensayos clínicos acerca de que el efecto del tratamiento sobre la variable sustituta corresponde al efecto sobre la variable clínica. La interpretación de un ensayo que utiliza como base una variable subrogada debe ser hecha en el contexto del número de individuos en riesgo y considerar las conclusiones con mucha precaución.
Resultados múltiples y análisis por subgrupo
Resultados múltiples
Aunque comúnmente es usado como variable respuesta primaria un evento único en los ensayos clínicos, en muchos otros la variable respuesta es una combinación de diversos eventos o se requieren múltiples mediciones de condiciones inherentes al tratamiento de prueba (otros serían múltiples comparaciones de tratamientos, mediciones repetidas, análisis por intervalos), incluyendo desde características clínicas como morbilidad, síntomas, calidad de vida, efectos colaterales del tratamiento, hasta utilización y costos de los servicios de salud. Es muy importante mencionar en el protocolo la pre-especificación de prioridades en respuestas múltiples.
Tradicionalmente se recomienda que sea diseñada en el protocolo una variable resultado primaria y las otras variables definirlas como secundarias o terciarias. El ensayo es, bajo estas condiciones, ponderado y monitoreado sobre las bases de una variable de resultado primaria. Una variable primaria global sería la mortalidad por todas las causas vasculares; se analizarían separadamente como variables secundarias los eventos que las formaron: infarto al miocardio, infarto cerebral, trombosis mesentérica, etcétera). El agrupamiento de los eventos debe hacerse de acuerdo con los objetivos del diseño para no mezclar eventos de mortalidad, morbilidad o calidad de vida.
Si se utilizan múltiples resultados primarios, se requiere de un número mayor de métodos estadísticos para contabilizar la multiplicidad y mantener al mismo tiempo los niveles de error tipo I en 0.0545 (esto quiere decir que se requieren ajustes del nivel a, valor de p, valores críticos para multiplicidad y pruebas globales que producen un índice resumido de la combinación de las variables resultado). Un método ampliamente usado es el de Bonferroni en el cual el nivel de significancia para cada variable resultado es a/k, donde k es igual al número de variables resultado, pero esta función es limitada si las variables de resultado están correlacionadas. Otras propuestas se han descrito para evitar problemas como el ordenar los valores univariados de p del menor al mayor y comparar cada valor de p con los niveles de a progresivamente mayores desde a/k, a/(k-1), hasta que a no rechace la hipótesis nula.46
Otros métodos de pruebas para ajustar el valor crítico de multiplicidad han sido desarrollados para tomar en consideración la distribución conjunta de las pruebas estadísticas de las múltiples variables resultado;47 mientras que Mantel48 sugiere un ajuste menos conservador: 1-(1-a), y Tukey49 remplaza 1/k con  cuando las variables resultado están correlacionadas, pero el grado de correlación se desconoce. Estimaciones globales de la medición pueden realizarse a través de los métodos de O'Brien50 en los cuales incluye métodos de suma de rangos y de regresión.
cuando las variables resultado están correlacionadas, pero el grado de correlación se desconoce. Estimaciones globales de la medición pueden realizarse a través de los métodos de O'Brien50 en los cuales incluye métodos de suma de rangos y de regresión.
Otros métodos propuestos para analizar resultados de múltiples variables son el uso de componentes principales y de análisis factorial.51
Análisis por subgrupo
Frecuentemente, la variable primaria está relacionada con otras variables de interés en las cuales puede ser importante evaluar el efecto del tratamiento al interior de subgrupos de sujetos. Tales características pueden ser la edad, el género, o se deben a que los sujetos fueron tratados en diferentes centros (ensayos multicéntricos). Otra razón puede ser probar la consistencia interna de los resultados entre las diferentes categorías. Cualesquiera que pueda ser la razón, es común dividir a los grupos aleatorizados en categorías o subgrupos para comparaciones de la intervención al interior de ellos. Es preferible que el establecimiento de los subgrupos haya sido definido por anticipado en el protocolo, así como identificadas las variables que se espera vayan a influir sobre el resultado primario, ya que de esta manera se puede considerar en el análisis (el prestablecimiento de los subgrupos y la preidentificación de variables) como una herramienta para mejorar la precisión y compensar por cualquier falla en el balance entre los grupos de tratamiento.
La confiabilidad de este análisis es pobre debido a problemas de multiciplicidad y a que los grupos generalmente son pequeños, en comparación con la muestra completa; por este motivo, el número de subgrupos debe mantenerse en un mínimo posible y preespecificarse en el protocolo como medida de incremento en la credibilidad de los resultados, pues el manejo de la hipótesis refleja un conocimiento biológico a priori más que una argumentación de resultados a posteriori. Por todos los motivos expuestos, los resultados de un análisis de subgrupos deben interpretarse de acuerdo con un análisis exploratorio, generador de hipótesis y nunca como un resultado definitivo. Estas recomendaciones aparentemente fáciles de extrapolarse a los diseños, no han sido del todo aceptadas y análisis e interpretaciones inapropiadas de resultados siguen ocurriendo como lo muestran Brookes y colaboradores52 en su estudio de simulación para cuantificar el riesgo de mala interpretación de resultados en este tipo de análisis. Los autores encontraron que con un poder nominal de 80% sólo obtuvieron oportunidad para detectar 29% de interacción en el efecto global del estudio. Al interior de los grupos sólo detectaba entre 7 y 64%, lo que lleva a interpretaciones erróneas de la efectividad subgrupal de un tratamiento y no en un efecto global de los mismos (mayor riesgo de falsos negativos). En algunos casos para detectar la interacción con un poder significativo tuvieron que inflar el tamaño de la muestra cuatro veces más.
Por último, en este análisis, las técnicas más utilizadas comprenden regresión lineal, árboles de regresión y estimaciones bayesianas en caso de requerirse ajuste de sesgos.
Resultados por tipo de comparación
Estudios de superioridad
En un ensayo clínico, la eficacia es el parámetro que demuestra superioridad de un tratamiento activo sobre el placebo o por mostrar un efecto dosis-respuesta. Los ensayos de superioridad intentan demostrar que la intervención experimental es mejor o igual que la intervención de control. Por ejemplo, para enfermedades graves, cuando un tratamiento ha mostrado ser eficaz por estudios de superioridad, proponer un nuevo ensayo clínico controlado con placebo puede ser considerado como no ético. La nueva terapia debe ser más fácil de administrar, mejor tolerada, menos tóxica o menos costosa, pero el beneficio intercambiado no debe reflejarse en renunciar al efecto del tratamiento.
Estudios de no inferioridad
En contraste con los estudios de superioridad, estos ensayos pretenden mostrar que la intervención experimental no es peor que el tratamiento estándar (pero no pretende mostrar superioridad) con algún margen de indiferencia. Adicionalmente, los investigadores deben demostrar que el nuevo tratamiento es mejor que el placebo en caso de que el ensayo tenga un brazo placebo. La conducción de un análisis de este tipo debe ser de muy alta calidad para detectar diferencias significativas; asimismo, debe tener un efectivo control de la intervención, sin olvidar que el margen de indiferencia no se basa en criterios estadísticos sino en la importancia médica del tratamiento en el contexto de intercambio riesgo/beneficio. El análisis estadístico de estos estudios se basa generalmente en el uso de los intervalos de confianza tal y como se muestra en la figura 6.
Citemos el caso del estudio OPTIMAAL53 que comparó losartan con captopril en pacientes con insuficiencia cardiaca. Su objetivo fue demostrar que losartan sería superior a captopril o por lo menos no sería inferior, basados en que podría ser mejor tolerado que captopril. La mortalidad resultó en 1.126 (IC 95% superior de 1.28), así que ni la superioridad ni la no inferioridad se lograron. Los investigadores entonces usaron datos históricos de pacientes tratados con captopril (RR=0.806, comparado con placebo) y lo multiplicaron con los resultados de OPTIMAAL (1.126*0.806) aproximándose al efecto de losartan con placebo si éste hubiera sido usado (el RR fue de 0.906), pero los resultados deben ser interpretados con extrema precaución a pesar de haber sido conducido muy bien el estudio OPTIMAAL.
Métodos estadísticos
Diversos diseños de análisis se han utilizado para evaluar las diferencias existentes entre los grupos sometidos a un tratamiento nuevo para compararlo con un placebo o un tratamiento convencional. La selección apropiada del análisis y la pre-especificación en el protocolo de estudio es un reto que muchos investigadores no toman en cuenta. Perrone54 realizó un estudio en 145 ensayos clínicos fase II en cáncer de mama y encontró resultados interesantes: 64.8% de ellos no tenía un propuesta estadística identificable, siendo los predictores más importantes para que identificaran un método estadístico definido (en modelos de regresión logística) el apoyo de una agencia financiadora, un único agente experimental y que el estudio fuera desarrollado como multicéntrico.
Los métodos de análisis estadísticos han evolucionado en años recientes, desde el tradicional análisis de supervivencia con sus estimaciones, hasta modelos más complejos vistos en los incisos previos, sin olvidarnos del análisis por riesgos competitivos (no censura el evento, por ejemplo, muerte por otras causas, sino que toma en consideración su incidencia acumulada),55 sin embargo, en este documento daremos una perspectiva de las técnicas más usadas en el desarrollo de los ensayos clínicos.
Descripción de los grupos de comparación
Un primer análisis que se efectúa en los ensayos clínicos controlados es comparar los grupos de tratamiento en sus condiciones basales y que generalmente son características demográficas (por ejemplo: edad, género), medidas antropométricas (ejemplo, masa corporal) condiciones inherentes a su estado clínico (ejemplo, severidad de enfermedad, control metabólico,) y otras variables pronósticas relacionadas con la variable resultado primaria. Generalmente presentadas como medidas de resumen (medias, medianas) y dispersión (desviaciones estándar, rangos) en variables continuas y en porcentajes para las variables categóricas. Esta comparación también es útil para describir la muestra de sujetos que entraron al estudio. Se han descrito pruebas de significancia (usualmente prueba de t y Ji cuadrada) con valores de p. Cuando ocurren diferencias entre los grupos de tratamiento, no necesariamente son debidas a falla en la aleatorización, sino por accidente. Altman56 recomienda que se comparen basalmente los descriptores usando una combinación de conocimiento clínico y sentido común. Si los grupos no están balanceados se debe hacer un análisis no ajustado y otro ajustado por la variable que desbalanceó los grupos.
El método estadístico usado para comparar dos medias en los diferentes grupos de tratamiento es la prueba de t de Student. Esta prueba compara la media de dos grupos de variables continuas y expresa la probabilidad de que cualquier diferencia sea debida al papel del azar (acepta la hipótesis nula) o que las diferencias son reales (rechaza la hipótesis nula). Dos supuestos básicos tiene la prueba de t: a) que los datos en ambos grupos siguen una distribución normal y, b) que para muestras no pareadas la varianza para cada grupo es igual. Cuando estos supuestos no se cumplen se recomienda el uso de pruebas no paramétricas. Cuando la variable tiene valores binarios (ejemplo, curación o muerte) se puede calcular la proporción del evento para cada grupo de tratamiento. El estadístico más común es la prueba de Ji cuadrada (Ji cuadrada o c2). La Ji cuadrada determina el número esperado del evento en ambos brazos de los grupos de tratamiento y los compara con los eventos observados, se expresa de la siguiente manera: c2= S (O — E)2 / E, donde O representa las frecuencias observadas y E representa las frecuencias esperadas en cada celda de una tabla de contingencias de 2 x 2. Esta prueba puede usarse para comparar más de dos proporciones. Un ejemplo de su uso se muestra en el cuadro IX.

Análisis de supervivencia
Es el método estándar para analizar el tiempo que transcurre entre un evento inicial (que determina la inclusión del individuo en el estudio) y un evento final (genéricamente llamado falla) que ocurre cuando el individuo presenta la característica para terminar el estudio (muerte, alta de la enfermedad, etc.). Se trata de un método flexible y puede medir múltiples eventos por sujeto, así como tomar en consideración en el análisis la información completa y parcial cuando así lo determina la ocurrencia de censuras (administrativas, por término del estudio o logro del número de sujetos requeridos, y no administrativas, por pérdidas en el seguimiento). El estimador de Kaplan-Mier58 toma en cuenta estas censuras estimando las tasas de supervivencia entre los grupos de tratamiento, asumiendo que la censura es no informativa (esto es, que las censuras no administrativas no están relacionadas con la ocurrencia del evento en estudio). Para medir diferencias en las curvas de supervivencia entre los grupos de comparación se toma en consideración los criterios de la prueba de Mantel y Haenszel (llamada también prueba de Long-Rank)59 quien pondera cada evento por igual y está cercanamente relacionada con las pruebas estadísticas del modelo de Cox.60 Este modelo de Cox o de riesgos proporcionales, ofrece la ventaja de contabilizar las variables basales y pronósticas y su estimador (función de hazard) es la tasa instantánea de morir para aquellos sujetos que sobrevivieron hasta el tiempo t. Este modelo especifica cómo cambia la función de riesgo básica (individuos con nivel de covariables cero) respecto de aquellos con covariables distintas de cero. Este cambio lo especifica el parámetro asociado a cada factor introducido al modelo y se interpreta como el cambio esperado en el cociente de riesgos entre un individuo en la población básica y uno fuera de ella, asumiendo que la razón de riesgos, para cualquier variable X, es constante a través del tiempo. Otro supuesto para cumplir con los modelos es que la curva de supervivencia de un grupo debe estar siempre por encima de la curva de supervivencia del otro grupo; en otras palabras, éstas no se pueden cruzar. El supuesto de proporcionalidad se evalúa mediante: a) las líneas del gráfico de las curvas de supervivencia (éstas no deben cruzarse), y b) las líneas del gráfico loglog: Ln[-Ln(S)] vs tiempo para todos los grupos (las líneas deben ser aproximadamente paralelas).
Si los supuestos del modelo son violados, deben entonces probarse modelos estratificados o buscar interacción.
Otros métodos de análisis
Regresión
El análisis de regresión mide la fuerza de asociación de una relación entre dos variables cuando una depende de la otra, bajo el supuesto de que esta relación es lineal. Si el análisis requiere que más variables sean estudiadas se puede extender el modelo a un análisis de regresión lineal múltiple.
Cuando el análisis es requerido a usar datos longitudinales, modelos de regresión mixtos pueden ser utilizados especialmente porque pueden incorporar efectos fijos o aleatorios y permiten determinar diferencias en las estructuras de covarianza. Estos modelos son una extensión de los modelos de regresión lineal para variables continuas y se representa: Yi= XIb + Zibi + ei, donde Yi es el vector de respuesta para el sujeto iésimo sobre el periodo puntual del seguimiento, Xi es la matriz del diseño usual para modelos fijos (tales como el tratamiento, variables basales, etc.) con su correspondiente coeficiente de regresión b, Zi es la matriz del diseño para efectos aleatorios (tal como un individuo tiende a desviarse de la media) con su correspondiente coeficiente bi y ei, es el vector de los residuales. Una regresión aleatoria permite a cada sujeto tener su propia tendencia en el tiempo y el efecto del tratamiento puede causar diferencias en la pendiente sobre el tiempo.
Cuando la variable resultado tiene estimaciones no normales se pueden utilizar modelos lineales generalizados y algunas extensiones de ellos.61
Existen aun más métodos de análisis que no fueron comentados en este capítulo y que serían motivo de un capítulo específico dentro de los que destacan meta-análisis, riesgos competitivos, análisis por intervalos, métodos bayesianos, análisis multinivel, análisis de covarianza, etcétera. Esta perspectiva presentada nos da una idea de cómo han evolucionado los análisis de los ensayos clínicos controlados llegando a una sofisticación única, pero que no rebasa la experiencia del científico que utiliza métodos tradicionales efectivos, plasmados en la planeación del diseño del estudio.
Etica de la investigación en ensayos clínicos aleatorizados
Proteger los derechos y el bienestar de los que participan en investigaciones científicas constituye el propósito actual de la ética de la investigación. Lamentablemente, la historia ha señalado investigaciones que no consideraron a los participantes como personas, sino como objetos de estudio. Por esta razón, la Declaración de Helsinki,22 en 1964, estableció uno de los primeros antecedentes en principios éticos de investigaciones en seres humanos. A este respecto, el Artículo 5º expresó su preocupación por el bienestar de los seres humanos, el cual debe tener [siempre] supremacía sobre los intereses de la ciencia y de la sociedad. En el cuadro X se describe el desarrollo histórico en las guías internacionales para la investigación con seres humanos.

Consentimiento informado (y genuino)
Un ensayo clínico exitoso depende, entre muchos factores, del proceso de obtención del consentimiento informado (CI). La tensión generada entre la necesidad de seleccionar a los participantes y la obligación de ofrecer ciertos tipos de protección ha hecho del CI un persistente reto ético ya que, (en los países en desarrollo, por ejemplo), las costumbres, las tradiciones y las concepciones que tienen acerca de la salud y la enfermedad pueden variar significativamente, además del posible desconocimiento relacionado con la investigación biomédica. De acuerdo con estándares internacionales, el CI consiste en una decisión de participar en una investigación hecha por un individuo competente que ha recibido la información necesaria, la ha comprendido adecuadamente y, después de considerar la información, ha llegado a una decisión sin haber sido sometido a coerción, intimidación ni a influencias o incentivos indebidos.23 Idealmente, la decisión de participar en un ensayo es un proceso que incluye la discusión del estudio con el investigador principal (IP) así como otros colaboradores, la garantía de que la información fue comprendida por el participante para que, finalmente, otorgue la firma en el documento de CI, ya sea el sujeto o su representante legal, aspectos que han sido estandarizados por organizaciones internacionales, como se describe en el cuadro XI.
Comités de ética
Universalmente, se acepta que no debe hacerse investigación médica en personas sin seguir los postulados y lineamientos nacional e internacionalmente aprobados. En México, la Ley General de Salud en Materia de Investigación,62 contempla en su Artículo 99, que toda institución de salud en donde se realice investigación para la salud deberá tener un Comité de Etica (CE) en el caso de que realice investigación en seres humanos. De acuerdo con las Guías Operativas para CE que evalúan investigación biomédica, propuestas por la Organización Mundial de la Salud (OMS), el propósito de un CE es contribuir a salvaguardar la dignidad, derechos, seguridad y bienestar de todos los y las participantes actuales y potenciales de la investigación.63
Los CE existen para asegurar, en primer lugar, que la investigación propuesta responda a las necesidades de salud de la población; en segundo lugar, que no exponga a los participantes a riesgos inaceptables e innecesarios y, en tercer lugar, que los participantes potenciales tendrán la garantía de ser completamente informados y, por lo tanto, tener la capacidad para evaluar las consecuencias previstas de su participación y decidir entonces su ingreso al estudio, mediante un consentimiento genuino.64
Conferencia internacional de armonización y buenas prácticas clínicas
Las Buenas Prácticas Clínicas (BPC), en inglés, Good Clinical Practice, es un estándar para el diseño, conducción, desarrollo, monitoreo, auditoría, registro, análisis y reporte de estudios de investigación clínica en los que participan seres humanos como sujetos de estudio. El cumplimiento de la BPC asegura dos aspectos fundamentales en los estudios en donde se pone a prueba un medicamento o un equipo de diagnóstico previo a su comercialización: a) la protección de los derechos, la seguridad, y el bienestar de los sujetos de estudio, y b) la credibilidad de los datos generados, necesaria para el registro de una innovación terapéutica en cualquier parte del mundo.
Los lineamientos para la conducción de estudios clínicos, conocidos como ICH (por sus siglas en inglés) constituye el estándar bajo el cual se rigen los países originalmente incluidos para llevar a cabo investigación en seres humanos y, por lo tanto, sugiere que cualquier grupo de investigación de otro país que desee realizar estudios clínicos y aportar datos que sean válidos en Estados Unidos de América (EUA), la Comunidad Europea y Japón, debe acogerse a estos lineamientos.65
Las recomendaciones de ICH están divididas en cuatro categorías, con temas específicos en cada una: Q, para Calidad, S para Seguridad, E para Eficacia y M para temas multidisciplinarios. En la sección ICH de eficacia se encuentran todos los temas relacionados con aspectos clínicos.
Buenas prácticas clínicas
Los lineamientos de las BCP son el estándar que se sigue durante el desarrollo de proyectos de investigación clínica. A este respecto, se encuentran claramente especificadas las responsabilidades y funciones del investigador y del patrocinador, los contenidos básicos del protocolo de investigación del ensayo clínico, el manual del investigador y los documentos esenciales para la conducción de un estudio clínico. Aquí se cubren todos los aspectos de preparación, monitoreo, reporte y archivo de estudios clínicos e incluye los lineamientos de BPC de la Unión Europea, Japón, EUA, Australia, Canadá, los Países Nórdicos, y la OMS, que deben ponerse en práctica cuando se generan datos clínicos que se pretenda someter a autoridades regulatorias o en cualquier proyecto de investigación clínica que pueda tener un impacto en la seguridad y bienestar de los sujetos participantes.
Responsabilidades del investigador
El investigador principal es por definición el encargado de vigilar el cumplimiento de los lineamientos de ICH, del respeto a las leyes locales y del apego al protocolo. Debe organizar un equipo de trabajo que le ayude a cumplir con las expectativas en cuanto a número de pacientes reclutados, tiempos para hacerlo y calidad de los datos generados. En el anexo I se describen las responsabilidades básicas del investigador principal.
Responsabilidades del patrocinador
El patrocinador es responsable de implementar y mantener sistemas para un aseguramiento de la calidad y control de calidad con procedimientos estándar de operación escritos para asegurar que los estudios sean conducidos y los datos sean generados, documentados (registrados) y reportados en cumplimiento con el protocolo, las BPC y los requerimientos regulatorios que apliquen (anexo II). Todos los acuerdos establecidos por el patrocinador con el investigador/institución y con cualquier otra parte involucrada en el estudio clínico deberán ser por escrito. El programa de control de calidad implementado por el patrocinador debe incluir actividades tanto en el ámbito interno como externo, asegurando que se encuentren correctamente documentados todos los procedimientos que tienen que ver con el estudio. Dicho programa debe incluir visitas regulares de monitoreo a los centros de investigación, detección oportuna de errores en la conducción del estudio y corrección inmediata de los mismos, con estrategias que aseguren su prevención en el futuro, incluyendo entrenamiento y reentrenamiento en caso necesario.
Monitoreo como parte de las responsabilidades del patrocinador
Uno de los elementos más importantes de la investigación clínica es la supervisión y doble verificación de los datos generados. El trabajo del monitor clínico tiene el objetivo primordial de cuidar que los derechos y el bienestar de los seres humanos estén protegidos, al mismo tiempo que vigila que los datos reportados del estudio estén completos, sean precisos y se puedan verificar de los documentos fuente. Todo esto, de acuerdo con el protocolo, con la BPC y con los requerimientos regulatorios aplicables.
De acuerdo con los lineamientos de ICH, monitoreo es "el acto de vigilar el proceso de un estudio clínico y asegurarse de que éste sea conducido, registrado y reportado de acuerdo con el protocolo, procedimientos normatizados de operación, la buena práctica clínica (BPC) y los requerimientos regulatorios aplicables".
Manejo de la información
La información es en realidad el producto final que se persigue con la conducción del estudio clínico. Los datos generados durante el estudio deben tener la calidad que se busca y estar disponibles en los tiempos requeridos para su análisis. Desde su inicio, todo estudio clínico tiene un plan de manejo de datos que implica tiempos establecidos previamente para su procesamiento, en donde se incluye captura, limpieza y análisis de la información.
Cuando se utilicen sistemas de manejo electrónico de datos el patrocinador deberá asegurar y documentar que el sistema esté diseñado para permitir cambios en los datos, que puedan ser documentados y que no se borren los registros originales. Además, debe haber un sistema de seguridad que impida el acceso no autorizado a los datos y mantener su respaldo adecuado.
Conclusiones
Los ensayos clínicos aleatorizados son los estudios más cercanos al método experimental, por lo que forman el paradigma de la investigación epidemiológica. Representan el diseño con el nivel más alto de causalidad, donde el investigador tiene control sobre la exposición, son prospectivos, tienen la ventaja de que en teoría se pueden evitar sesgos, tienen un elevado nivel de comparabilidad de poblaciones (confusión) y de información. Su práctica, sin embargo, es muy compleja y en países en desarrollo se debe priorizar, además de los aspectos metodológicos, la garantía de asegurar los derechos de los pacientes.
Referencias
1. Packard FR. Life and times of Ambroise Pare, 1510-1590. Nueva York (NY): Paul B. Hoeber; 1921.
2. Good P. A manager's guide to the designs and conduct of clinical trials. Hoboken (NJ): Wiley and Sons; 2002:47-64.
3. Greeno C. Major alternatives to the classic experimental design. Fam Process 2002;41:733-736.
4. Barbui C, Violante A, Garattini S. Does placebo help establish equivalent in trials of new antidepressants? Eur Psychiat 2000;15: 268-273.
5. Rivera JA, Sotres-Alvarez D, Habicht JP, Shamah T, Villalpando S. Impact of the Mexican program for education, health, and nutrition (Progresa) on rates of growth and anemia in infants and young children: A randomized effectiveness study. JAMA 2004;291:2563-2570.
6. Montgomery AA, Peters TJ, Little P. Design, analysis and presentation of factorial randomised controlled trials. BMC Med Res Methodol 2003;3:1-5.
7. Menéndez C, Kahigwa E, Hirt R, Vounatsou P, Aponte J, Font F et al. Randomised placebo-controlled trial of iron supplementation and malaria chemoprophylaxis for prevention of severe anaemia and malaria in Tanzanian infants. Lancet 1997;350:844-850.
8. Mundorf TK, Ogawa T, Naka H, Novack GD, Crockett RS; US Istalol Study Group. A 12-month, multicenter, randomized, double-masked, parallel-group comparison of timolol-LA once daily and timolol maleate ophthalmic solution twice daily in the treatment of adults with glaucoma or ocular hypertension. Clin Ther 2004;26:541-551.
9. Traversa G, Bignami G. Ethics problems in phase IV of drugs studies. Ann Ist Super Sanita 1998;34:203-208.
10. Linden M. Differences in adverse drug reactions in phase III and phase IV of the drug evaluation process. Psychopharmacol Bull 1993;29:51-56.
11. Moher D, Schultz KF, Altman D. The CONSORT statement: Revised recommendations for improving the quality of reports of parallel-groups randomised trials. JAMA 2001;357:1191-1194.
12. Naylor CD, Guyatt GH. Users' guides to the medical literature. X. How to use an article reporting variations in the outcomes of health services. JAMA 1996;275:554-558.
13. Begg CB, Cho MK, Eastwood S, Horton R, Moher D, Olkin I et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276:637-639.
14. Schulz K, Grimes D. Blinding in randomised trials: Hiding who got what. Lancet 2002;359:696-700.
15. Emanuel EJ, Miller FG. The Ethics of placebo-controlled trials. A middle ground. N Engl J Med 2001;345:915-918.
16. Schafer A. The ethics of the randomized clinical trials. N Engl JMed 1982;307:719-724.
17. Freedman B. Placebo-controlled trials and the logic of clinical purpose. IRB 1990;12:1-6.
18. Hrobjartsson A, Gotzsche PC. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. N Engl J Med 2001;344:1594-1602.
19. Kienle GS, Kiene H. The powerful placebo effect: Fact or fiction? J Clin Epidemiol 1997;50:1311-1318.
20. Beecher HK. The powerful placebo. JAMA 1955;159:1602-1606.
21. Rothman KJ, Michels KB. The continuing unethical use of placebo controls. N Engl J Med 1994;331:394-398.
22. World Medical Association. (1996, 2000). Declaration of Helsinki. Edinburgh, Scotland. Disponible en: http://www.wma.net/e/policy/17-c_e.html. [2004 septiembre 10].
23. Council for International Organizations of Medical Sciences, 1993; 2002. International Ethical Guidelines for Biomedical Research Involving Human Subjects. Geneva, Switzerland. 2002 revision. Disponible en: http://www.cioms.ch. [2004 septiembre 10].
24. Karrison TG, Huo D, Chappell R. A group sequential, response-adaptive design for randomized clinical trials. Control Clin Trials 2003;24:506-522.
25. Deeks JJ, Dinnes J, D'Amico R, Sowden AJ, Sakarovitch C, Song F et al. Evaluating non-randomised intervention studies. Health Technol Assess 2003;7(27):iii-x,1-173.
26. Krause MS, Howard KI. What random assignment does and does not do. J Clin Psychol 2003;59:751-766.
27. Berger VW, Bears JD. When can a clinical trial be called "randomized"? Vaccine 2003 17;21:468-472.
28. Soares I, Carneiro AV. Intention-to-treat analysis in clinical trials: Principles and practical importance. Rev Port Cardiol 2002;21: 1191-1198.
29. Scott NW, McPherson GC, Ramsay CR, Campbell MK. The method of minimization for allocation to clinical trials. A review. Control Clin Trials 2002;23:662-674.
30. Schulz KF, Grimes DA. Allocation concealment in randomised trials: Defending against deciphering. Lancet 2002;359:614-618.
31. Slack MK, Draugalis JR. Establishing the internal and external validity of experimental studies. Am J Health Syst Pharm 2001;58:2173-2181.
32. Altman DG, Schulz KF. Statistics notes: Concealing treatment allocation in randomised trials. BMJ 2001;323:446-447.
33. Cotton PB. Randomization is not the (only) answer: A plea for structured objective evaluation of endoscopic therapy. Endoscopy 2000;32:402-405.
34. Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ 1999;319: 670-674.
35. Zelen M, Lee SJ. Models and the early detection of disease: Methodological considerations. Cancer Treat Res 2002;113:1-18.
36. Friedman LM, Furberg CD, DeMets DL. Fundamentals of clinical trials. 3rd. ed. New York (NY): Springer-Verlag; 1998.
37. Peduzzi P, Henderson W, Hartigan P, Lavori P. Analysis of randomized controlled trials. Epidemiol Rev 2002;24:26-38.
38. Green SB. Design of randomized trials. Epidemiol Rev 2002;24:4-11.
39. The Anturane Reinfarction Trial. Sulfinpyrazone in the prevention of cardiac death after myocardial infarction. N Engl J Med 1978;298: 289-295.
40. The Anturane Reinfarction Trial Research Group. Sulfinpyrazone in the prevention of sudden death after myocardial infarction. N Engl J Med 1980;302:250-256.
41. Temple R, Pledger GW. The FDA's critique of the anturane reinfarction trial. N Engl J Med 1980;303:1488-1492.
42. Influence of adherence to treatment and response of cholesterol on mortality in the coronary drug project. N Engl J Med 1980;303: 1038-1041.
43. DeMets DL. Statistical issues in interpreting clinical trials. J Intern Med 2004;255:529-537.
44. Wang D, Bakhai A, Maffulli N. A primer for statistical analysis of clinical trials. Arthroscopy 2003;19:874-881.
45. Zhang J, Quan H, Ng J, Stepanavage ME. Some statistical methods for multiple endpoints in clinical trials. Control Clin Trials 1997;18:204-221.
46. Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat 1979:65-70.
47. Westfall PH, Young SS. P-value adjustments for multiple tests in multivariate binomial models. J Am Stat Assoc 1989;84:780-786.
48. Mantel N. Assessing laboratory evidence for neoplastic activity. Biometrics 1980;36:381-399.
49. Tukey JW, Ciminera JL, Heyse JF. Testing the statistical certainty of a response to increasing doses of a drug. Biometrics 1985;41:295-301.
50. O'Brien PC. Procedures for comparing samples with multiple endpoints. Biometrics 1984;40:1079-1087.
51. Henderson WG, Fisher SG, Cohen N, Waltzman S, Weber L. Use of principal components analysis to develop a composite score as a primary outcome variable in a clinical trial. The VA Cooperative Study Group on Cochlear Implantation. Control Clin Trials 1990;11:199-214.
52. Brookes ST, Whitely E, Egger M, Smith GD, Mulheran PA, Peters TJ. Subgroup analyses in randomized trials: Risks of subgroup-specific analyses; power and sample size for the interaction test. J Clin Epidemiol 2004;57:229-236.
53. Dickstein K, Kjekshus J. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: The OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan. Lancet 2002;360: 752-760.
54. Perrone F, Di Maio M, De Maio E, Maione P, Ottaiano A, Pensabene M et al. Statistical design in phase II clinical trials and its application in breast cancer. Lancet Oncol 2003;4:305-311.
55. Sylvester R, Van Glabbeke M, Collette L, Suciu S, Baron B, Legrand C et al. Statistical methodology of phase III cancer clinical trials: Advances and future perspectives. Eur J Cancer 2002;38(Suppl 4):S162-S168.
56. Altman DG. Comparability of randomized groups. Statistician 1985;34:125-136.
57. Lazcano-Ponce EC, Sloan NL, Winikoff B, Langer A, Loggins C, Heimbenger A et al. The power of information and contraceptive choice in a family planning setting in Mexico. Sex Transm Infect 2000;76: 277-281.
58. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958;53:457-481.
59. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966;50:163-170.
60. Cox DR. Regression models and life tables. J R Stat Soc, series B, 1972; 34:187-220.
61. Breslow NR, Clayton DG. Approximate inference in generalized linear mixed models. J Am Stat Assoc 1985;88:9-25.
62. Secretaría de Salud. Reglamento de la Ley General de Salud en Materia de Investigación para la Salud. [1986]. Disponible en: http://www.salud.gob.mx/unidades/cdi/nom/compi/rlgsmis.html. [2004 septiembre 10].
63. Gilbert C, Fulford KW, Parker C. Diversity in the practice of district ethics committees. BMJ 1989;299:1437-1439.
64. Torgerson DJ, Dumville JC. Ethics review in research: Research governance also delays research. BMJ 2004;328(7441):710.
65. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) adopts Consolidated Guideline on Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use. Int Dig Health Legis 1997;48:231-234.
Solicitud de sobretiros
Dr Eduardo Lazcano Ponce
Centro de Investigaciones en Salud Poblacional
Instituto Nacional de Salud Pública
Avenida Universidad 655, colonia Santa María Ahuacatitlán
62502, Cuernavaca, Morelos, México
Correo electrónico: elazcano@correo.insp.mx
Fecha de recibido: 20 de septiembre de 2004
Fecha de aprobado: 23 de septiembre de 2004
* Nota: el diseño factorial en este caso hace la doble aleatorización en un solo tiempo de manera tal que cuando se forman los cuatro grupos la doble aleatorización ya fue llevada a cabo. Así se garantiza que los sujetos estén potenciados por el doble proceso. Pero el procedimiento se lleva a cabo en un solo tiempo antes de la formación de los grupos.



{kind=link}
{kind=link}
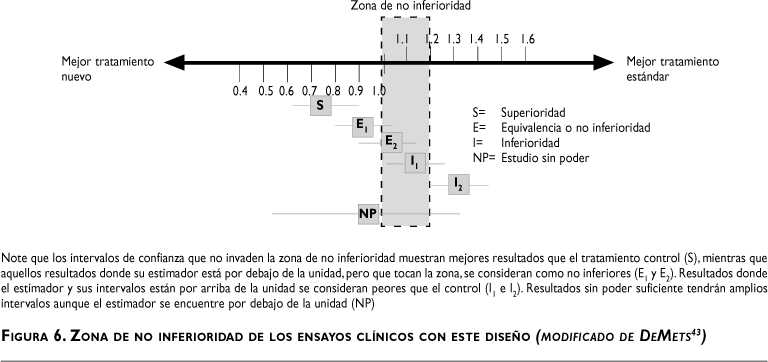{kind=link}
{kind=link}
{kind=link}