











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. INTRODUCTION
Infrared (IR) spectroscopy is a useful tool in studying systems at temperatures around 102 K. Objects at these temperatures such as protoplanetary, accretion, and protostellar disks, significantly radiate in this region of the electromagnetic spectrum. Commonly, IR studies are conducted in ground-based observatories; thus, the observed spectrum presents lines of both the astronomical source and the terrestrial atmosphere. Therefore, including the atmospheric absorption is a crucial step towards any study involving IR astronomical spectra in order to avoid misidentifications with the atmospheric absorption lines. This implies dealing with the atmosphere, which is made up of a large quantity of gas molecules at temperatures of the same order as those of the observed sources.
The Earth’s atmosphere is mostly composed of N2 (78.09 %) and O2 (20.95 %), both accounting for almost 99% of the average composition of the dry atmosphere (Cox 2000). These homonuclear3diatomic molecules are inactive in the IR because of their null electric dipole moment. In fact, according to Gross selection rules (Atkins & de Paula 2014), fundamental vibrational transitions are forbidden for these molecules and no IR contribution is observed. On the other hand, atmospheric heteronuclear molecules such as CO, CO2 and H2O, are some of the ones responsible for the major features of the IR absorption atmospheric spectrum, due to rotational and vibrational transitions.
The spectrum theory of diatomic molecules is presented in detail in classical books (Kronig 1930; Herzberg 1950) and also in physical chemistry texts (Atkins & de Paula 2014). As the atmosphere is composed of diatomic and polyatomic gases, aerosols, dust, pollutants and other constituents, studying its spectrum is a complex task. The variable concentration of CO and CO2 due to its major sources: anthropogenic, biological, oceanic, combustion, photochemical, and the strongly variable concentration of the water vapor due to atmospheric and oceanic conditions, cause the IR spectrum of the terrestrial atmosphere, called the telluric4 spectrum, to change in time scales of seconds and up to months. Currently, simulating the atmospheric molecular transitions requires the development of complex algorithms5 which consider the different quantum interactions present in these molecules (Rudolf et al. 2016; Ulmer-Mollet al. 2019, and references therein).
The aim of the present work is to obtain, as a pedagogical exercise, analytical expressions for the IR transition energies of heteronuclear atmospheric molecules using undergraduate quantum mechanics, and to show that these transitions produce bands in the IR spectra. The organization of this study is as follows: in § 2 we present the Hamiltonians that describe the simplest approximation to the vibrational and rotational transitions of molecules. In § 3 we compute vibration and rotation transitions, using a more realistic Hamiltonian through a Morse potential. In § 4 we present our results, showing that CO IR transitions form bands. Finally, in the last section we give the main conclusions of this work.
2. MODELING VIBRATION AND ROTATION OF DIATOMIC MOLECULES
To describe the energy spectrum of a diatomic molecule the vibrational and rotational degrees of freedom are considered. Studies of degrees of freedom considering complex models are discussed by Stuart (2004). In this section, we present the vibrational and rotational Hamiltonians for a system without and with an interaction term between each degree of freedom.
2.1. Vibrational Hamiltonian
A diatomic gas molecule has a spectrum originated in part by the vibrational
motion. Because these vibrations are not completely harmonic, there must be
introduced correction terms that account for the non-harmonic behavior of the
molecule. As a first approximation, one can add to the potential an extra term
proportional to
where µ is the reduced mass of the system, ω is the oscillation frequency and G 1 is a small parameter related to the first correction of the potential. Higher order correction terms are described by the factors Gi>1.
Remembering the results for the regular quantum harmonic oscillator, we use the
creation â, annihilation â † and number
For the anharmonic Hamiltonian, the term corresponding to
Hence, the cubic term given by equation (1) can be rewritten in the following way:
To continue, the action of the correction term over | ni is obtained:
Given the small size of the correction G 1 term, perturbation theory can be applied. Notice that this Hamiltonian does not present a first order correction to its energy levels. We invite the readers to calculate the second order perturbation theory correction (see Cohen-Tannoudji et al. 1977) to find the following results for the energy:
(2) Plugging back these correction terms into the corrected energy term, a new energy spectrum is found.
This Hamiltonian describes the degrees of freedom corresponding to the vibrational nature of molecules.
2.2. Rotational Hamiltonian
When a molecule rotates, the centrifugal force pulls the atoms apart causing an increase in its moment of inertia and decreasing the rotational constant associated to this energy term. This effect couples both types of interactions, vibration and rotation. In order to understand this interaction, the following analogy can be useful: a pair of masses (atoms of a diatomic molecule) are attached at the ends of a spring, so that the masses can vibrate along the line between them. The spring is fixed at some point to the center of a rotating disk. The system has no friction. Due to rotation, the Coriolis force deflects the masses, changing the vibration frequency of the spring, reflecting an interaction between rotation and vibration in this mechanical system. This is analogous to the interaction between rotation and vibration in diatomic molecules.
Returning to the effect of the centrifugal force on the coupling of the rotation and vibration interactions and, therefore, on the rotational Hamiltonian, a term named centrifugal diatomic distortion is defined as
The r variable describes the inter-nuclear distance between the atoms composing the molecule. The rotational Hamiltonian is now the following:
where L is the angular momentum operator ( 𝐿 = 𝑟 × 𝑝 ) and I is the moment of inertia. The corrected Hamiltonian in equation (4) shares the same eigenstates | l,mi that are used in the rotational uncorrected Hamiltonian, where D = 0. Thus the action of the Hamiltonian over these eigenstates produces:
3. HAMILTONIANS FOR DIATOMIC MOLECULES
3.1. Model 1: Combined Hamiltonian
Before using the vibration and rotation Hamiltonians given by equations (1) and
(4), it is necessary to find an appropriate basis which diagonalizes the
Hamiltonians in the corresponding Hilbert space.If these Hamiltonians are
defined as
This description supposes that the movement of the rotation and the vibration are described in the same coordinates where the Hamiltonians commute. If the vibrations were considered along a different direction, these would affect the separation radius of the molecule, changing the moment of inertia of the system. These corrections cannot be ignored and correction terms must be added. This coupling is studied in the following section.
By taking into account both interaction terms, rotation and vibration, a net
Hamiltonian can be studied. This Hamiltonian is fully described by the quantum
numbers n and l. This fact is a consequence of the commutation relation
Tables 1 and 2 give the vibrational and rotational transitions for an initial state | n,li. The energy transitions displayed above only show a change of state in a single Hamiltonian (vibrational or rotational). All the other transitions are described by the combination of transitions between both quantum numbers given by the selection rules ∆n = ±1,±2 and ∆l = ±1,±2.
TABLE 1 ALLOWED TRANSITIONS FOR VIBRATIONAL HAMILTONIAN
|
|
|
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
TABLE 2 ALLOWED TRANSITIONS FOR ROTATIONAL HAMILTONIAN
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Notice that each initial state | n,li has 24 possible final states given the corrections proposed by this model. It is worth mentioning that this correction is very restrictive as the rotation is fully confined to one direction. Moreover, the energy constant associated with this rotational degree of freedom is greater than that found in any other direction.
3.2. Model 2: Morse Potential
The Morse potential is commonly used to describe the vibrational behavior of diatomic molecules. Its asymptotic behavior, given by the maximum energy of vibrations (the dissociation energy, D E , related to the depth of the potential well) more accurately describes the physics of diatomic molecules than the quantum harmonic oscillator.
In the above expression, r0 is the equilibrium bond distance and a is a constant related to the strength of the bond. By using this expression, the Schr¨odinger equation can be perturbatively solved obtaining the following n-dependent energy spectrum
3.3. Rotation-Vibration Interaction
In § 3.1 we have described the energy spectrum of a diatomic molecule when both vibrations and rotations are present in the system. In such a way, the Hamiltonian can be split into two non-interacting terms, which share a common basis. However, this is not the general case as vibrations can affect the way the rotational spectrum is defined. This effect can be understood by considering the moment of inertia of a single molecule: given the vibration of the molecule, the length of the bond separating the molecules will change. Therefore, in order to introduce the radial separation (r), the constants B and D appearing in the rotational Hamiltonian are now considered as functions of this variable:
Under the assumption that the vibration frequency is much greater than the rotational frequency, the average separation radius can be computed, and the effective separation can be described. To compute this effective separation, we consider the average radius in the wave function describing the oscillatory movement:
Following the same procedure, the quantities B(r) and D(r) are defined through their mean values in a full period of oscillation:
Through this approximation, one obtains the following new constants related to the unperturbed terms, B e and D e , presented in § 2.2 for the rotational Hamiltonian:
where the subindex e refers to the constants given in the equation (4). The new constants, α e and β e , are the ones which introduce the interaction of these two Hamiltonians. These values have been reported by Le Floch (1991).
3.4. Rotational and Vibrational Hamiltonian with Morse Potential
To better describe the energy spectrum of a diatomic molecule, we should use a
total Hamiltonian that includes a term related to the Morse potential (given in
equation 8) and other term related to the rotational and vibrational coupling
(involving the expressions given in equation 13). Obtaining this total
Hamiltionian,
where the first two terms are the given by equation (9) and the last two terms are the rotationalvibrational energy discussed in § 3.3. Reordering terms, we obtain:
where ω χ = ω2(4D E )−1. In the expression above, the first term corresponds to the energies of the harmonic oscillator and rigid rotor, the second one reflects corrections to the energy given by anharmonicity and centrifugal distortion, and the last term corresponds to rotation-vibration interaction.
As an exercise, we propose that readers work on some of the following ideas that could improve the presented models:
-Include higher order correction terms in the anharmonic oscillator. (These corrections make possible several other transitions given by the selection rules).
-Include higher order corrections in the rotational Hamiltonian, which would have the same effect as the corrections of the anharmonic oscillator.
-Include molecular interactions, which would take place in a diatomic gas.
4. DISCUSSION
In the previous section we presented a model considering rotational and vibrational Hamiltonians with a Morse potential as a first approximation for diatomic molecules without molecular interactions. Using equation (14) with the transition values ∆n and ∆l of 0,±1,±2, we obtained the wavelengths corresponding to IR transitions. Our calculations, in spite of not having the amplitude of probability for every transition, expose the complexity of the IR spectrum. We have used equation (14) rather than equation (7) since this model does not include the mixed energy terms.
We apply the results found to the CO molecule, as shown in Figure 1. Molecular constants for the CO were taken from Le Floch (1991) and are reported in Table 3. Experimental measurements of these constants using a near-IR spectrophotometer were presented by Mina-Camilde et al. (1996).
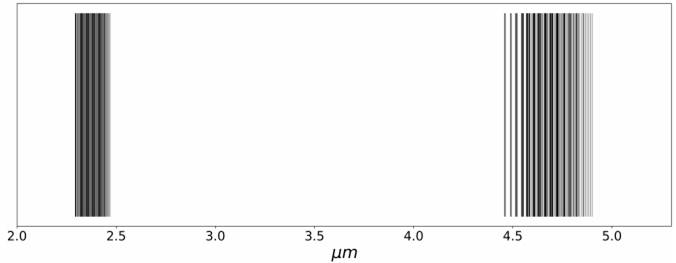
Fig. 1 Computed IR spectrum of the CO molecule. Fundamental and first overtone transitions are present around 2.35µm and 4.67µm, respectively. Bands are clearly appreciated.
TABLE 3 MOLECULAR CONSTANTS FOR CO
|
|
|
|
| 0.193128081434 | 0.6121593342 | 0.1750399404 |
|
|
|
|
| -0.9316227 | 2169.81259421 | 13.287834373 |
The CO spectrum shown in Figure 1 illustrates that IR transitions form bands. Our model illustrates how complex it is to obtain the infrared spectrum of heteronuclear diatomic molecules: every transition will depend on the initial state of vibration and rotation, i.e., both quantum numbers n and l. Note that the parameters used in the Hamiltonians depend on the moment of inertia, mass and internuclear distance and on the existence of mixed terms in the energy given by equation (15).
We compare our theoretical CO IR wavelengths with those of the high resolution (R = 40000) telluric spectrum6 created from data made available by the NSO/Kitt Peak Observatory, in the K S − and M−bands.
Although the atmosphere spectrum presents multiple absorption lines of CO, CO2 and H2O, we have identified several CO lines in the observed telluric spectrum using our computed CO lines as the reference spectrum. This identification was produced by means of the IRAF7 task identify of the package onedspec (devoted to reduction and calibration of spectra) reaching an average precision of 0.003 per cent with respect to our theoretical wavelengths. We also made a linear regression between our wavelengths and those identified in the telluric spectrum. The slope of the regression is equal to one, the intercept is zero (with a precision of 10−7) and the R2 coefficient is one (with a precision of 10−11). This linear regression shows that our computed CO wavelengths agree with those identified in the telluric spectrum.
Figure 2 illustrates the K S − and M− bands IR telluric spectra, where CO absorption lines are clearly identified with our computed lines. In these regions the absorption features present in the atmosphere are caused by water vapour, carbon dioxide and greenhouse gases.
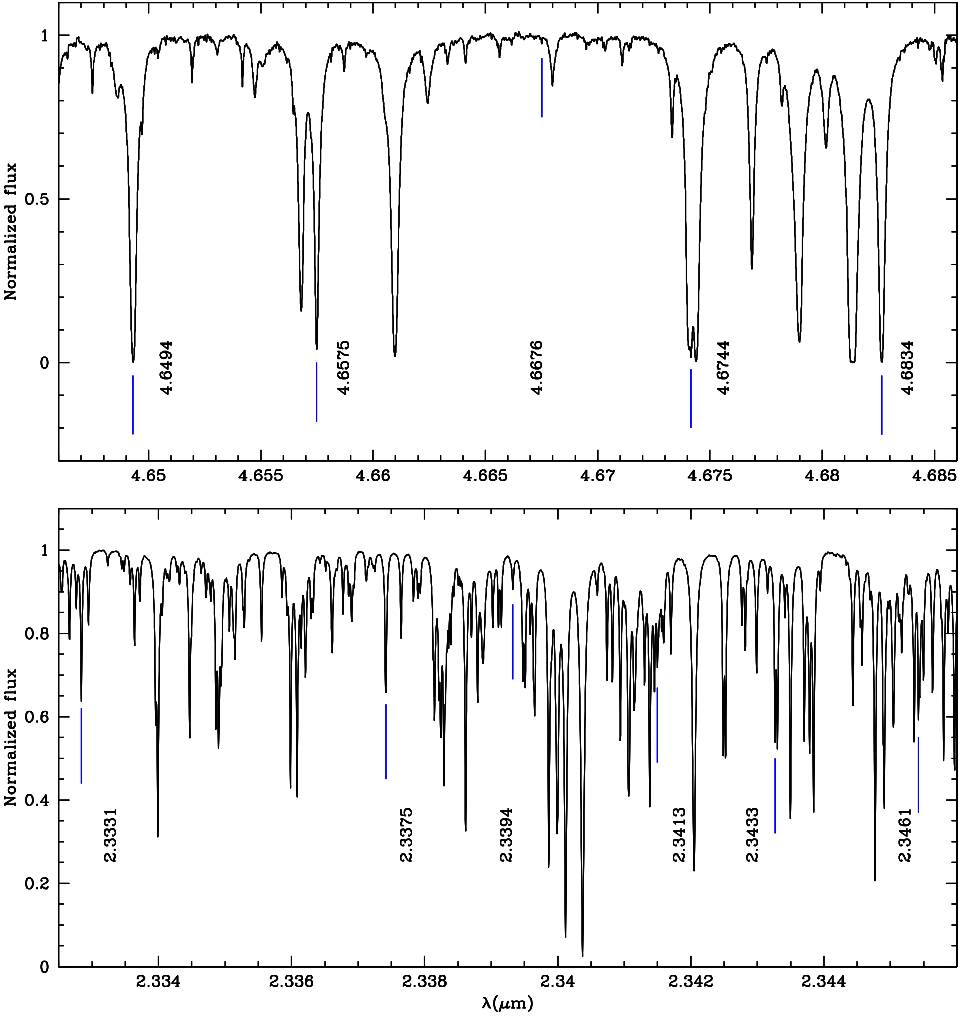
Fig. 2 Line identification. K S − and M−band telluric spectra (black) are shown in the bottom and upper panels, respectively. In both panels we show the CO absorption lines identified with our computed wavelengths (blue). Fundamental mode transitions occur in the K S −band. First overtone transitions are present in the M−band. The color figure can be viewed online.
5. CONCLUSIONS
In order to analyse the IR spectra of astronomical sources it is important to identify the telluric features that are produced by the terrestrial atmosphere. In this work we present a quantum mechanics model, useful to obtain analytical expressions for the transition energies of heteronuclear atmospheric molecules. The Hamiltonian used includes the rotation and vibration of diatomic molecules, as well as the interactions between them. We showed that it is possible, with this model, to identify transitions of CO molecule that are present in the K S − and M− bands.

