











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkThe development of affordable “new generation” sequencing methods and the bioinformatic capacity to analyze the large amounts of data obtained, have unleashed the potential to carry out detailed genetic analysis in non-model organisms which were previously inaccessible. Our model of study is Agave tequilana, however the strategy we used can be applied to most organisms including plant, fungal, bacterial or insect species. Although A. tequilana is an important crop in Mexico predicted to generate around 6 billion dollars in profit annually by 2024 from tequila production, the perennial, monocarpic life cycle of this species, the practice of removing immature inflorescences in the field and the strict control by the Tequila Regulatory Council over the germplasm grown have led to a situation where a single cultivar (A. tequilana Weber var. azul) is reproduced asexually and grown over 95,000 hectares in the 5 Mexican states authorized under “Denomination of Origin” for tequila production. Very few breeding programs have been implemented in agave species and no improved cultivars have been developed either for yield in terms of sugar production, control of flowering or resistance to pests and pathogens. Disease and pest resistance are of particular importance given the practice of exploiting a single genotype over thousands of hectares making agave plantations extremely vulnerable to attack. Our initial omics analyses have focused on unraveling the genetics of sugar (fructan) metabolism and control of flowering in A. tequilana.
Strategy
For many crop plants such as corn, beans, tomato (Hirsch and Buell, 2013) among others, the determination of a whole genome sequence in order to carry out genetic analysis has been feasible, however, A. tequilana has a genome content of almost twice the size (4000Mb) (Palomino et al., 2007) of corn and development and construction of a whole genome sequence is still technically challenging. Therefore, our initial strategy was to develop methods for transcriptome (RNAseq) analysis in agave species. Transcriptome analysis offers the advantages that less data needs to be generated and processed lowering costs. Additionally, data can be related to tissue types or developmental stages providing information on expression levels of specific genes under specific conditions. We have generated transcriptome data for A. tequilana, A. deserti, A. striata and A. victoria-reginae (Ávila de Dios et al., 2015) and using this information have identified, cloned and characterized the cDNAs (coding sequences) responsible for the regulation of fructan metabolism and some members of a family of genes involved in the regulation of flowering time in A. tequilana.
A total of 15 genes involved in fructan/sucrose metabolism were identified and classified as encoding fructan synthesizing enzymes, fructan degrading enzymes or invertases. Genetic transformation of agave species is still laborious and inefficient and therefore we are using heterologous systems such as P. pastoris and A. thaliana in order to functionally characterize the identified genes/enzymes. In silico expression patterns based on transcriptome data indicate which genes are expressed in specific tissues or during specific developmental stages indicating their relative importance in different processes or metabolism. Based on these data we have determined and compared expression patterns for each of the fructan metabolism genes and most of these in silico expression patterns have now been confirmed by qRT-PCR (Ávila de Dios et al., 2019, Pérez-López et al., 2021).
By employing a similar strategy, genes from several families known to be involved in the regulation of flowering time in the model system A. thaliana (FT, gibberellin metabolism, MYB factors) were also identified in A. tequilana and based on in silico expression patterns a working model for regulation of flowering in A. tequilana was proposed (Ávila deDios et al., 2019). Currently we are functionally characterizing in detail the A. tequilana FT family.
An example of preliminary omics analysis in Colletotrichum lindemuthianum
The C. lindemuthianum - common bean pathosystem involves a gene-for-gene interaction between resistance (R) genes in the common bean host cultivars and avirulence (avr) genes in the fungal pathogen. Previous work in our group identified many distinct C. lindemuthianum races based on the differential series of common bean cultivars (González et al., 1998, Mendoza et al., 2001, Rodríguez-Guerra et al., 2003, González-Chavira et al., 2004). We initiated a transcriptome strategy to compare the differential gene expression patterns at the early stages of infection on the common bean cultivar BAT93 inoculated with either a virulent or a non-virulent race (Race 1088 and Race 256 respectively) of C. lindemuthianum (Figure 1 and Table 1). Details of the experimental strategy are shown in Table 2. RNAseq was carried out on samples from each time point (samples from 4-96 hours were pooled) (Table 3) and the data was analyzed and compared in order to eventually identify genes expressed differentially during compatible and incompatible interactions (Medina-Chavez and Simpson unpublished).
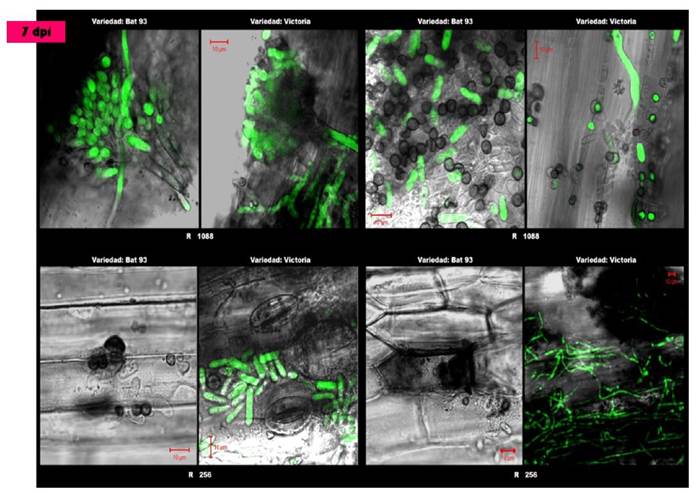
Figure 1 Microscopic analysis 7 days after inoculation of common bean seedlings with Colletotrichum lindemuthianum. In the upper left quadrant, the formation of reproductive structures (acervuli) by the pathogen is observed in the compatible interactions for both cultivars with strain 1088. On the right side for the same strain a large number of conidia and appresoria are shown. In the interactions with strain 256, a large number of hyphae are observed for the cultivar Victoria as well as the presence of young conidia on the necrotic tissue of the plant, whereas for the cultivar BAT 93, hypersensitive reactions caused by the defense response are observed with the localized death of cells in contact with the pathogen.
Table 1 Reactions of BAT93 and Victoria to inoculation of C. lindemuthianum Races 256 and 1088.
| Cultivar | Race | Reaction | Infection level |
|---|---|---|---|
| BAT93 | 1088 | Compatible | 4 |
| Victoria | Compatible | 4 | |
| BAT93 | 256 | Incompatible | 0 |
| Victoria | Compatible | 4 |
Genomic and transcriptomic analyses are currently essential and cost-effective tools for genetic analysis and can easily be applied to plants and/or their pathogens. The P. vulgaris-C. lindemuthianum pathosystem is particularly amenable given the well -documented series of differential cultivars and corresponding C. lindemuthianum races. The recent release and ongoing analysis of several P. vulgaris genomes (Rendón-Anaya et al., 2017) will greatly facilitate this research.
Table 2 Summary of samples taken for RNAseq analysis.
| Compatibility | Sample time 15 min | Compatibility | Sample time 15 min | Compatibility | Sample time 15 min |
|---|---|---|---|---|---|
| Incompatible | 30 min | Compatible | 30 min | Un-inoculated | 30 min |
| 1 h | 1 h | 1 h | |||
| 2 h | 2 h | 2 h | |||
| 4 h | 4 h | 4 h | |||
| 8 h | 8 h | 8 h | |||
| 24 h | 24 h | 24 h | |||
| 48 h | 48 h | 48 h | |||
| 72 h | 72 h | 72 h | |||
| 96 h | 96 h | 96 h |

