











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El sarcoma de Ewing es la segunda neoplasia ósea maligna más frecuente en pediatría; sin embargo, también puede presentarse en tejidos blandos.1 Los tumores neuroectodérmicos primarios son tumores de células pequeñas y redondas derivadas del tejido blando, pertenecen a la familia del sarcoma de Ewing.2,3 Estas neoplasias exhiben un fenotipo neural, expresan la proteína MIC2- (CD99) y 85% de los casos muestra translocación cromosómica t(11:22) (q24; q12).4,5 El tumor neuroectodérmico primario puede presentarse en órganos sólidos como riñón, pulmón, ovario, vejiga y páncreas, entre otros.5,6 El tumor neuroectodérmico primario del páncreas es un tumor muy poco frecuente que habitualmente se presenta en niños o adultos jóvenes.7-9 En la literatura sólo se han reportado 17 casos.9-11 En este artículo se presentan las características clínicas, el tratamiento y la evolución de un paciente pediátrico con tumor neuroectodérmico primario del páncreas, así como una revisión de la literatura.
PRESENTACIÓN DEL CASO
Niña de 9 años de edad proveniente del estado de Tabasco. Inició su padecimiento con ictericia, palidez, astenia, adinamia, anemia, pérdida de peso de 3 kilogramos en tres meses y dolor abdominal epigástrico, por lo que se inició protocolo de estudio realizando una tomografía y una resonancia magnética que mostraron un tumor retroperitoneal de 14 x 10 x 8 cm, localizado en la cabeza del páncreas. Se tomó una biopsia per-cutánea que reportó una neoplasia maligna de células pequeñas y se le envió a nuestro centro, el Instituto Nacional de Pediatría. A su ingreso se encontró, en la exploración física, dolor epigástrico de tipo cólico de intensidad variable, irradiado a la región lumbar, hepatomegalia de 3 cm por debajo del borde costal, melena, sin evidencia de tumores o ganglios. Se hicieron los siguientes estudios de laboratorio: bilirrubina total: 0.26 mg/dL; aspartato-aminotransferasa: 74 UI/L; alanina-aminotransferasa 121 UI/L, fos-fatasa alcalina 245 UI/L, lactato-deshidrogenasa 208, gamma-glutamiltranspeptidasa 196 UI/L, amilasa 1.1 UI/L.
Se le practicó cirugía de Whipple con resección completa de un tumor en la cabeza del páncreas, de 14 x 10 x 8 cm en sus diámetros mayores y 270 g de peso (Figura 1), que infiltraba al duodeno, grasa y ganglios linfáticos peripancreá-ticos; así mismo se realizó colecistectomía. La cirugía tuvo una duración de 7 horas. No hubo complicaciones transoperatorias y el sangrado quirúrgico fue mínimo. La paciente ingresó estable a la unidad de cuidados intensivos.

Figura 1 Pieza quirúrgica producto de la resección de un tumor en la cabeza del páncreas y segmento de duodeno; dimensiones 14 x 10 x 8 cm; peso 270 g.
El reporte histopatológico confirmó un tumor neuroectodérmico primitivo/sarcoma de Ewing, en la cabeza del páncreas, con menos de 10% de necrosis y calcificaciones focales, con infiltración al duodeno, conducto de Wirsung y tejidos blandos peripancreáticos, así como a uno de cuatro ganglios locorregionales. No se encontró actividad en los márgenes quirúrgicos de la resección (Figuras 2-4 (3)). Al quinto día de poso-peratorio la paciente desarrolló choque séptico, insuficiencia renal y salida de material fecaloide por la herida quirúrgica, por lo que tuvo que ser sometida a una segunda laparotomía por sepsis abdominal donde se evidenció dehiscencia de la anastomosis yeyuno-pancreática. Se lavó la cavidad, se realizó necrosectomía y una nueva anastomosis con colocación de drenajes. Posteriormente la paciente tuvo fístula biliocutánea que fue tratada con sistema de presión negativa y drenaje percutáneo, así como tratamiento médico con octreotida y tratamiento médico de la sepsis. La paciente requirió antibióticos de amplio espectro y la complicación se resolvió.

Figura 2 Fotomicrografía: ácinos pancreáticos (izquierda) en íntima relación con una neoplasia de células pequeñas redondas y azules (derecha).


Figura 4 Fotomicrografía: características morfológicas. Células pequeñas redondas y azules (izquierda) positivas para la tinción de inmunohistoquimica CD99 (derecha); junto con otras tinciones apoya el diagnóstico de tumor neuroectodérmico primitivo/sarcoma de Ewing.
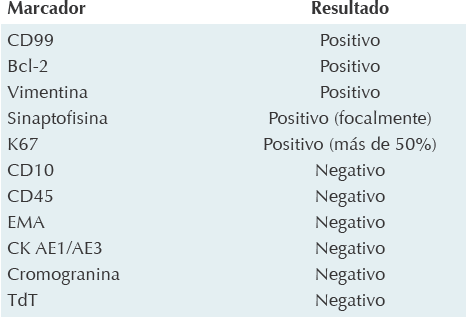
En el tumor primario se realizaron los marcadores de inmunohistoquímica referidos en el Cuadro 1. Como parte de la extensión de la neoplasia maligna se realizaron los siguientes estudios para evaluar la extensión de la enfermedad, establecer un plan de tratamiento y hacer un pronóstico: tomografía de pulmón de alta resolución, gammagrama óseo y biopsia bilateral de médula ósea, éstos se reportaron negativos para metástasis.
Tres meses después de la cirugía, y una vez que se habían resuelto las complicaciones quirúrgicas, se inició el tratamiento de quimioterapia con base en el Protocolo Nacional para sarcoma de Ewing con ciclofosfamida (2.1 gr/m2 al día durante dos días), vincristina (2 mg/m2 por 1 día), doxorrubicina pegilada (50 mg/m2 por 1 día) alternado con etopósido (100 mg/m2 al día durante 5 días) e ifosfamida (2 g/m2 al día durante 5 días) por 8 ciclos y radioterapia en hemiabdo-men superior, 50.4 Gy en 30 sesiones.11 Durante el tiempo que la paciente recibió quimioterapia presentó 4 cuadros de neutropenia y fiebre, los cuales se resolvieron sin complicaciones. Actualmente la paciente se encuentra bajo vigilancia, desde marzo del 2013, en buenas condiciones generales y sin evidencia de actividad tumoral.
ANÁLISIS CUALITATIVO DE LA LITERATURA
Se realizó una búsqueda exhaustiva en las siguientes bases: Medline 1950-2013 (OVID), Embase 1980-2013 (OVID), LILACS (1998 a 2013), ARTEMISA (1999 a 2013) y SCIELO (1999 a 2013). Se encontraron 9 artículos en los cuales se reportaban casos de pacientes con tumor neuroectodérmico primitivo/sarcoma de Ewing en páncreas. Movahedi, en 2002, reportó 7 casos, sin embargo, sólo uno se presentó en población pediátrica. De los 16 casos encontrados sólo 7 fueron en pacientes pediátricos con una media de edad de 10.8 años. Se describen las características clínicas, tratamiento y evolución de los pacientes en el Cuadro 2.

DISCUSIÓN
El sarcoma de Ewing es el segundo tumor maligno primario de hueso más común en pediatría, precedido por el osteosarcoma. Sin embargo, el sarcoma de Ewing también puede afectar tejidos blandos.2,7 La histología característica de estos tumores son células pequeñas, redondas y azules; es común en niños, adolescentes y adultos jóvenes.1,5 Se considera que no hay diferencia histológica entre los tumores de esta familia.2,8
Los tumores de páncreas son extremadamente raros en niños.7,5 Los tumores neuroectodérmicos primitivos son histológicamente indistinguibles de los del tipo óseo y se han documentado en otros sitios como vagina, septo retrovaginal, intestino delgado, ovarios, próstata, esófago y riñón.5,3 En la literatura médica internacional hay muy pocos casos registrados. En 2013 se habían reportado sólo 17 casos en los que se describían las manifestaciones más comunes al momento del diagnóstico.4 En 85% de los casos el dolor abdominal fue el signo más importante de la enfermedad; con menor frecuencia se reportaron vómito, fiebre y pérdida de peso, como en el caso de la paciente que presentamos. Sólo se reportaron 7 casos pediátricos (en menores de 18 años).
El tratamiento de estos pacientes no está bien establecido debido a la baja frecuencia de la enfermedad. De los casos reportados 14.2% fueron tratados únicamente con cirugía, 14.2% con quimioterapia y radioterapia y 57.1% con quimioterapia y cirugía, dependiendo de la extensión de la enfermedad. En uno de los casos no se reportó el tratamiento utilizado. En todos los casos el tratamiento inicial fue la cirugía de Whipple (pancreatoduodenectomía). Internacionalmente, en pacientes con sarcoma de Ewing sin especificar su localización, se recomienda el uso de quimioterapia con las mismas drogas que empleamos en el Protocolo Nacional, basado en el protocolo Euroewing 99, que ha mostrado ser bien tolerado y logra una buena respuesta.11 Nuestra paciente recibió tratamiento con vincristina, ciclofosfamida y doxorrubicina, alternado con ifosfamida y etopósido; además de radioterapia abdominal por enfermedad ganglionar. Este tratamiento ha sido bien tolerado y se ha reportado supervivencia de hasta 70% de los casos en series internacionales. De los 7 pacientes pediátricos reportados en la literatura 5 (71.4%) se encuentra vivos. Nuestra paciente está viva y sin datos de actividad tumoral en un seguimiento que dura ya 48 meses.
Es importante considerar que 85% de los pacientes con sarcoma de Ewing presenta la translocación t(11,22)(q24;q12). En nuestra paciente no evaluamos esa alteración pero, actualmente, el diagnóstico integral de estos pacientes incluye la determinación de alteraciones cromosómicas, por lo que es necesario integrar esas pruebas al diagnóstico de los pacientes en México.

