











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Actualmente el tratamiento farmacológico de la obesidad y la resistencia a insulina, como antesala de diabetes mellitus tipo II y síndrome metabólico en niños y adolescentes, es más frecuente debido al fracaso de la terapia con ejercicio y dieta,1,2 a los malos hábitos alimenticios, al bullicio excesivo y ritmo de vida acelerado y violento de las ciudades y al sedentarismo imperante originado del uso indiscriminado de instrumentos que han hecho la vida más cómoda, que ha reducido al mínimo el gasto energético en las actividades más triviales de la sociedad actual.3 De ahí que la obesidad y sus alteraciones metabólicas se hallan en pediatría y, de manera alarmante, se espera que vayan en aumento. El hidrocloruro de metformina es uno de los fármacos que se ha utilizado frecuentemente para control de peso, resistencia a insulina y diabetes mellitus tipo II en niños y adolescentes,4-6 ya que contribuye a disminuir la absorción de glucosa desde el intestino hacia la sangre, inhibe la liberación de glucosa desde el hígado y hace que el músculo esquelético y el tejido adiposo respondan de forma más eficiente a la insulina;7,8 asimismo, contribuye a restablecer los lípidos a niveles normales, pues promueve la oxidación de ácidos grasos libres,8 sin causar hipoglucemia ni acidosis láctica.
La metformina tiene una biodisponibilidad de 50 a 60% por vía oral, su concentración plasmática máxima se alcanza a las 2.5 ± 1 horas y la vida media de eliminación a las 3.6 horas en población mexicana.9 Las tabletas de metformina disponibles comercialmente para adultos constan de 500, 850 y 1000 mg, tanto de la marca innovadora Glucophage, Roche®, como de las genéricas.4,10 Los pacientes de 12 a 16 años de edad que requieren tratamiento con metformina pueden tomar dosis similares a las de los adultos, dependiendo de su peso corporal; sin embargo, los adolescentes sin sobrepeso o los menores de 8 años que tienen estas mismas alteraciones metabólicas, requieren ajuste de dosis, adaptación para la deglución y mejoramiento del sabor a partir de las tabletas comerciales.
Los laboratorios farmacéuticos mexicanos no han puesto en el mercado una presentación pediátrica de metformina debido a que la población pediátrica es un mercado mucho menor que el de adultos y ésta requiere volúmenes más pequeños de producto y con mayor variedad de dosis, por las variaciones de peso y talla en este grupo de edad.11,12 Estos factores hacen poco costeable la producción de metformina pediátrica por la industria farmacéutica, ya que su uso en niños y adolescentes es menos frecuente que en adultos. Actualmente, el personal médico y los familiares deben fraccionar las tabletas para ajustar la dosis. Este procedimiento resuelve temporal y parcialmente el problema de dar el medicamento al niño; no obstante, origina dosis subterapéuticas, dificultad para administrar el medicamento y biodisponibilidad indeterminada.
Por otro lado, la determinación de niveles plasmáticos de fármaco es necesaria para supervisar el apego al tratamiento y correlacionar el éxito terapéutico. Esta actividad es especialmente importante tratándose de fármacos adaptados a la población pediátrica en formulaciones extemporáneas como la metformina, cuya presentación comercial está disponible únicamente para adultos.
La técnica de gota de sangre seca (DBS por Dried Blood Spots), desarrollada por Güthrie en 1963,13 ha sido propuesta en los últimos años para vigilancia clínica de fármacos, estudios farmacocinéticos (tanto en humanos como en animales), epidemiológicos, metabolómicos y toxicológicos.14-19 Esta técnica es muy poco invasiva ya que, sin necesidad de canalizar, se pueden obtener múltiples muestras sanguíneas de muy pequeño volumen, con la punción del talón o el dedo y se transfieren a papel filtro, donde se almacenan y se pueden analizar mucho tiempo después. Originalmente se planeó para detectar enfermedades metabólicas en el recién nacido.13
OBJETIVO
Comparar la farmacocinética de metformina en formulación líquida edulcorada sin calorías (con sucralosa a 1%) por vía oral, previamente desarrollada en nuestro laboratorio,20 con la de la tableta de 500 mg fraccionada a la mitad, en voluntarios adultos sanos, para demostrar que el comportamiento farmacocinético de la metformina tiene cambios insignificantes tras modificar su presentación.
MATERIAL Y MÉTODOS
En el presente estudio se implementó y validó un método analítico para determinar la concentración plasmática de metformina por técnica de gota de sangre seca y cromatografía líquida de ultradesempeño-espectrometría de masas, que tiene la ventaja de ser muy sensible y rápido, por lo que se empleó para determinar la biodisponibilidad de formulaciones extemporáneas de metformina, con potencial aplicación en pediatría, ya que no hay presentaciones comerciales disponibles, con excepción de Estados Unidos y Canadá, donde sí se comercializan. La formulación de metformina desarrollada en nuestro laboratorio corresponde a un apoyo a los clínicos del Servicio de Endocrinología, en donde se brinda atención médica a una gran cantidad de niños y adolescentes, principalmente de escasos recursos. Económicamente sería muy difícil que estos pacientes tuvieran que importar una presentación líquida de un medicamento que deben tomar crónicamente, por muchos años y en algunos casos de por vida.
Reactivos
Estándares puros de clorhidrato de metformina y de ranitidina, MP Biomedicals® (Fountain Pkwy, Solon OH, USA). Acetonitrilo y metanol grado HPLC, JT Baker. Acetato de amonio, Merck® (Darmstat, Germany). Ácido fórmico, Sigma Aldrich® (St. Louis MO, USA). Para todas las soluciones y diluciones se empleó agua bidestilada filtrada por sistema Milli-Q® (Millipore, Molsheim, France).
Condiciones cromatográficas
El sistema de cromatografía líquida de ultradesempeño, modelo AcQuity® (Waters Co., Milford MA, USA) acoplado a un espectrómetro de masas Quattro micro tándem® (Waters Micromass, Manchester, UK) operado en el modo de ionización positiva por rocío electrónico (ESI (+): "electrospray positive ionization mode") y controlado por el programa informático Mass Lynx® NT 4.0 (Waters Micromass, Beverly, MA, USA). La separación de los compuestos se llevó a cabo en una columna Acquity UPLC BEHHILIC, 2.1 × 100 mm, 1.7 μm (WatersTM) a 40°C, automuestreador a 15°C, velocidad de flujo de 0.25 mL/min. El tiempo de corrida fue de 3.5 minutos. Los tiempos de retención fueron 2.07 minutos para metformina y 2.51 minutos para ranitidina. La fase móvil consistió en acetato de amonio 0.5 M con acetonitrilo (ACN) en proporción 80:20, respectivamente.
Condiciones espectrométricas
Los analitos se midieron por supervisión de reacción múltiple usando las siguientes transiciones iónicas: m/z1+ 130 > 70.58 Th para metformina y 315.17 > 176.05 Th para ranitidina. La energía de cono fue de 20 y 25 V para metformina y ranitidina, respectivamente; mientras que la energía de colisión fue de 20 V para ambos. Se utilizó un "dwell" de 0.25 s. Los datos obtenidos se procesaron mediante el programa Mass Lynx® Ver. 4.1
Elaboración de estándares y controles
Las soluciones iniciales de metformina y ranitidina (1 mg/mL) se prepararon individualmente en metanol a 50% (v/v, en agua). De la solución inicial se hicieron diluciones en metanol 50% para obtener soluciones de trabajo (soluciones 20X) con concentraciones finales de 1,000, 4,000, 8,000, 12,000, 16,000 y 20,000 ng/mL para los puntos de la curva de calibración; así como 3,000, 10,000, y 15,000 para los puntos controles de calibración bajo, medio y alto. La curva de calibración se preparó mezclando 950 μL de sangre total (hematocrito 45%) con 50 μL de solución 20X de metformina a las diferentes concentraciones. 40 μL de esta mezcla se vertieron en tarjetas Whatman S&S 903 (tarjetas Güthrie) en cada círculo por separado. Las tarjetas se dejaron secar en posición horizontal por 12 horas a temperatura ambiente. Una vez secas se almacenaron debidamente etiquetadas en bolsas de plástico de baja permeabilidad a gases, acompañadas de material secante a 4°C hasta el momento de analizarse.
Curva de calibración
Constó de seis puntos: 50, 200, 400, 600, 800 y 1,000 ng/mL de acuerdo con las concentraciones plasmáticas informadas en niñas adolescentes hispanas.21 Los puntos controles bajo, medio y alto estarán representados por tres concentraciones intermedias a la curva de calibración que fueron: 150, 500 y 750 ng/mL.
Procesamiento de las muestras (extracción)
Se cortaron cinco discos de 3.0 mm de diámetro de cada tarjeta empleando una perforadora manual McGill®, Incorporated. La cantidad adecuada de discos se colocó en un microtubo de 1.5 mL y se les agregaron 750 μL de una mezcla de metanol y acetonitrilo en proporción 67:33. Se agitaron por dos minutos a temperatura ambiente en un agitador de toque (Vortex®) y luego se sonicaron por 5 minutos. Las muestras se centrifugaron a 12,000 rpm por 5 minutos a temperatura ambiente. Se colectó la fase orgánica y se transfirió a otro tubo, se evaporó a 40°C bajo corriente de oxígeno. La muestra extraída y seca se reconstituyó con 150 μL de una solución de ranitidinade25 ng/mL en agua: acetonitrilo, en proporción 70:30 (v/v). Para la detección se inyectaron 5 μL al sistema cromatográfico.
Validación del método
La validación del presente método se realizó con estricto apego a las normas oficiales mexicanas NOM-177-SSA1-2013 (FDA 2001, EMEA) y a su vez en acuerdo con lineamientos internacionales para métodos bioanalíticos.22 Las pruebas realizadas en cromatografía líquida de ultradesempeño y espectrometría de masas en tándem fueron: supresión iónica por efecto de matriz, selectividad del método para fármacos concomitantes (amoxicilina, ácido acetilsalicílico, captopril, glibenclamida, omeprazol y acetaminofén), acarreo, linealidad, precisión, exactitud, límite inferior de cuantificación, recuperación absoluta y estabilidad del fármaco en solución y extraído de la muestra a corto y largo plazos. Por cromatografía líquida de ultradesempeño con detección ultravioleta se determinaron la linealidad, precisión, exactitud, recuperación absoluta, límite de cuantificación y selectividad a los mismos fármacos concomitantes.
Población de estudio
El presente estudio es clínico de fase II. Se estudiaron 12 voluntarios adultos sanos, de edad promedio 24.3 ± 1.8 años, 7 varones y 5 mujeres, con índice de masa corporal de 24.9 ± 2.5 kg/ m2, estatura promedio de 1.68 m y peso corporal promedio de 71.3 ± 10.4 kg, que no estuvieron tomando ningún otro medicamento por al menos una semana antes, sin consumir alcohol ni tabaco los cinco días previos al estudio y que aceptaron participar voluntariamente mediante firma de consentimiento informado.
Se excluyeron a los voluntarios con antecedentes de toxicomanías, alteraciones hepáticas o renales, VDRL, hepatitis o VIH positivos, además de hipersensibilidad a las biguanidas, embarazo confirmado o lactancia. Ningún voluntario se eliminó del estudio. A cada voluntario se le informó cabalmente el procedimiento, el propósito y las ventajas del protocolo; firmaron una carta de consentimiento informado manifestando su autorización para participar.
Diseño del estudio
El presente estudio fue aprobado por los comités de Ética e Investigación del Instituto Nacional de Pediatría, con número de registro 100/2013. Se realizó con estricto apego a los principios éticos enunciados en la Declaración de Helsinki.
Estudio clínico, prospectivo, longitudinal, analítico, cruzado en dos, en 12 voluntarios adultos sanos
Una semana antes de la primera etapa los voluntarios acudieron a realizarse una historia clínica completa y una química sanguínea de 6 elementos en ayuno que incluyó glucosa, urea, creatinina, triglicéridos, transaminasas hepáticas y colesterol total, a fin de comprobar los correctos funcionamientos renal y hepático, así como para descartar que tuvieran diabetes mellitus II.
El ensayo se llevó a cabo en forma aleatoria. Al momento de firmar el consentimiento informado los voluntarios tomaron un sobre cerrado que contenía la formulación que tomarían primero. La secuencia aleatoria se generó mediante pequeños papeles que tenían escritos los números correspondientes a la fecha en que les tocaba tomar la tableta fraccionada o la formulación líquida. Los pacientes fueron ciegos a la elección de los sobres. El analista fue ciego al tipo de muestra y a la identidad del voluntario.
El estudio se llevó a cabo en dos etapas: previo ayuno de 8 horas desde la noche anterior al estudio, los voluntarios tomaron una dosis única que resulta prácticamente inocua al organismo. En la primera etapa 6 voluntarios tomaron 250 mg en 5 mL de la formulación extemporánea edulcorada de metformina (con sucralosa a 1%), elaborada a partir de las tabletas del innovador (Glucophage®, Roche®) y los otros 6 tomaron media tableta de 500 mg de metformina (250 mg) de la marca innovadora (Cuadro 1).
En la segunda etapa, después de un período de eliminación total del fármaco de una semana (periodo de lavado),18 los primeros 6 voluntarios tomaron la media tableta de metformina y los restantes tomaron la formulación extemporánea.
Cuadro 1 Datos demográficos de los voluntarios participantes en el estudio cruzado. Etapa I: del día 1 al día 30 el grupo 1 recibió metformina en formulación líquida; el grupo 2 recibió la tableta fraccionada. Etapa II: del día 37 al 67 se invirtieron las presentaciones farmacéuticas de metformina que tomaron ambos grupos

IMC: índice de masa corporal; DE: desviación estándar.
De este modo, cada voluntario fue su propio testigo. La solución de metformina se elaboró siempre nueva con cada voluntario y justo antes de tomarla. Se programaron tres voluntarios por día, a fin de ejecutar en tiempo y forma la toma y manejo de las muestras.
Durante todo el procedimiento el voluntario estuvo cómodo y tuvo las atenciones médicas debidas de nutrición y registro de signos vitales como tensión arterial, frecuencia cardiaca, frecuencia respiratoria y temperatura. A todos los individuos se les dio a beber 100 mL de una solución glucosada a 10%, a los 30, 90 y 120 minutos de haber administrado el medicamento. En caso de mostrar signos de hipoglucemia comprobada por Dextrostix®, se tuvo preparada una solución de 100 mL con glucosa a 20%. En ningún caso fue necesario administrarla.
Toma de muestras
A cada paciente se le recolectaron siete muestras de sangre por punción digital con lancetas activadas por contacto BD Microtainer® a las 0.25, 0.5, 1, 2, 4, 8 y 12 horas después de haber tomado la tableta fraccionada de metformina (250 mg) o bien la formulación edulcorada (250 mg/5 mL); dichas muestras se colectaron en forma de gota en tarjetas Güthrie, una tarjeta de 4 círculos por cada tiempo de muestreo y se almacenaron en bolsas de plástico herméticas con desecante a temperatura de 2 a 8°C hasta el momento de su análisis.
Parámetros farmacocinéticos
Se caracterizó el comportamiento farmacocinético de la metformina en formulación extemporánea, comparado con el de la tableta fraccionada. Los parámetros calculados fueron la concentración plasmática máxima (Cmáx), el tiempo en que ésta se alcanza (Tmáx), el tiempo de vida media de eliminación (t1/2), la constante de eliminación (ke), el volumen aparente de distribución (Vd), el aclaramiento (Cl), el área bajo la curva de la última concentración cuantificable (AUC0-12) y el área bajo la curva del tiempo cero hasta infinito (AUC∞). Se presentan los resultados promedio de todos los voluntarios, incluyendo la desviación estándar.
Análisis estadístico
Se realizó con estricto apego a la norma NOM177-SSA1-2013,21 mediante la prueba t doble unilateral (Schuirmann), considerando los datos obtenidos de la transformación logarítmica de AUC y Cmáx, y un intervalo de confianza a 90% para el cociente entre los promedios de los medicamentos de prueba (formulación extemporánea) y de referencia (tableta fraccionada). Se consideró un intervalo de confianza de 85 a 125% para los datos no transformados. El análisis de datos de la farmacocinética se efectuó mediante el programa Win Nonlin ver 3.1. habiéndose ajustado los datos a un modelo abierto de un compartimento.
RESULTADOS
Los datos demográficos de los voluntarios sanos se presentan en el Cuadro 1.
Validación del método analítico
El método desarrollado y validado en este trabajo para la cuantificación de niveles plasmáticos de metformina demostró ser lineal en el intervalo de concentraciones desde 50 hasta 1,000 ng/mL. La curva de calibración, para cuantificar la cantidad de fármaco de muestras problema, tuvo las siguientes concentraciones: 50, 200, 400, 600, 800 y 1,000 ng/mL, con un límite inferior de cuantificación de 50 ng/ mL (Cuadro 2). Se obtuvo un coeficiente de determinación (r2) de 0.998035. La variabilidad intrae interdía no fue considerable y el método fue preciso y exacto, con coeficientes de variación menores a 7.5% (Cuadro 3). Las muestras extraídas y almacenadas a 4°C fueron estables por 17 horas. Asimismo, estables hasta 15 horas en automuestreador (15°C) y hasta 2 meses a -80°C.
Cuadro 2 Linealidad
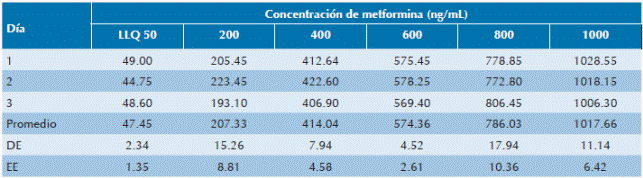
LLQ: límite inferior de cuantificación; DE: desviación estándar; EE: error estándar (DE/√n). Curva de calibración: y=0.0872219x+2.56985. Coeficiente de correlación: r=0.999017, r2=0.998035.
Cuadro 3 Parámetros de validación del método analítico* para cuantificar metformina en gotas de sangre seca

DE: desviación estándar. EE: error estándar (DE/√n). La cantidad calculada para los controles de calidad QC1, QC2 y QC3 fue 150, 500 y 750 ng/mL, respectivamente.
*Cromatografía líquida de ultradesempeño y espectrometría de masas en tándem.
El método fue selectivo para fármacos de posible prescripción concomitante, que fueron: captopril, ácido acetilsalicílico, orlistat, amoxicilina, glibenclamida, acetaminofén y omeprazol, este último fármaco aparece en el cromatograma de la metformina pero no interfiere con su análisis.
En la Cuadro 4 se describen las condiciones espectrométricas del método, que describen el protocolo de detección de metformina por cromatografía líquida de ultradesempeño y espectrometría de masas en tándem.

Biodisponibilidad relativa de metformina en formulación extemporánea líquida
La metformina en tableta tuvo una Cmáx de 553.4 ng/mL, mientras que en formulación líquida alcanzó un valor de 692.2 ng/mL. El valor Tmáx fue 12 minutos menor con la solución edulcorada (1.6 vs. 1.8 h). La ke fue menor con la forma líquida (0.29 vs. 0.32 h-1), el AUC0-12 y el AUC0-∞ fue igual en ambas formas (F=0.002, Fcrit=6.3) y (F=0.005; Fcrit=6.2) respectivamente, de acuerdo con el análisis de varianza de una vía (p ≤ 0.05) (Cuadro 5, Figura 1).
Cuadro 5 Farmacocinética comparada de metformina en voluntarios adultos sanos (n=12). Los datos se procesaron por análisis de varianza de un solo sentido (ANOVA, p ≤ 0.05)

El porcentaje de biodisponibilidad F(%) para AUC0-12 y AUC∞ fue de 1.01 (AUC0-12 de la formulación/AUC0-12 de la tableta; AUC∞ de la formulación/AUC∞ de la tableta).
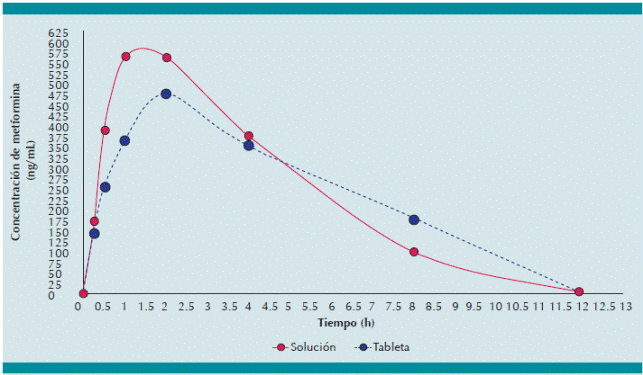
Figura 1 Comportamiento farmacocinético de la metformina en voluntarios adultos sanos (n=12) después de tomar metformina por vía oral (250 mg). Comparación entre la formulación líquida edulcorada y la tableta fraccionada.
Asimismo, no se presentó ningún indicio de reacciones adversas al fármaco en los voluntarios, de hecho no fue necesario administrarles solución glucosada a 20% en ningún momento del estudio.
DISCUSIÓN
En el presente estudio se determinó la biodisponibilidad relativa de la metformina en una formulación líquida, edulcorada con sucralosa a 1%, que es un endulzante sin calorías ("nonnutritive sweetener"). En estudios previos se ha demostrado que esta formulación desarrollada en nuestro laboratorio es estable fisicoquímicamente hasta por 30 días a temperatura ambiente y hasta 60 días en refrigeración; asimismo mostró estabilidad microbiológica, pues no se observó crecimiento de microorganismos mesófilos aerobios, enterobacterias ni hongos.20 Tanto la Agencia Europea de Medicamentos (EMA) como la Agencia de Alimentos y Medicamentos (FDA) de Estados Unidos establecen evaluar la biodisponibilidad de formulaciones extemporáneas en voluntarios adultos sanos y extrapolar los resultados a la población pediátrica, dado que no es ético ejecutar este tipo de estudios en niños sanos.12 En nuestro estudio ha quedado demostrado que no existen diferencias significativas entre los parámetros farmacocinéticos de la formulación comparados con los de la tableta comercial fraccionada. No se presentaron reacciones adversas a la formulación extemporánea de metformina.
La formulación líquida de metformina desarrollada en nuestro laboratorio20 contenía sucralosa a 10%, lo que le confirió sabor dulce y muy agradable. Sin embargo, en el presente estudio se decidió reducir su concentración a la mínima requerida para enmascarar el mal sabor del fármaco, a fin de evitar cualquier posible toxicidad causada por el edulcorante.23 Así, la formulación líquida administrada a los voluntarios tuvo sólo 1% de sucralosa. No obstante, 5 de los 12 voluntarios refirieron percibir un sabor desagradable.
A este respecto hay reportes que sustentan la seguridad e inocuidad del consumo de sucralosa en pacientes pediátricos y en la población en general. Los estudios del consumo de sucralosa en humanos, lactantes, niños, adultos, mujeres embarazadas, mujeres lactantes, diabéticos y obesos han mostrado que la ingestión diaria de sucralosa del triple de la dosis máxima, durante tres meses, no produce reacciones metabólicas adversas, no modifica los niveles de insulina plasmática ni los de glucemia cuando se ingiere de manera aislada; no altera los niveles de hemoglobina glucosilada a largo plazo, no causa toxicidad aún con el consumo de 18 kg acumulados durante la vida, no modifica la expresión ni la función de los receptores GLUT-2, por lo que no interfiere con la absorción intestinal de glucosa.24 De modo que en los próximos estudios, que involucran la evaluación de la biodisponibilidad en pacientes pediátricos, se retomará el diseño de esta formulación empleando 10% de sucralosa a fin de mejorar notablemente la palatabilidad y asegurar la aceptación de la dosis y la adherencia a su consumo por parte de la población pediátrica.
Método analítico
Es posible cuantificar metformina mediante el mismo proceso de extracción y determinación de la cromatografía líquida de ultradesempeño y espectrometría de masas en tándem. La supervisión terapéutica de fármacos es una manera de obtener información sobre la exposición del fármaco en el organismo de un individuo, de tal manera que podemos saber que tan eficaz o tóxica resulta su exposición. Rutinariamente, las concentraciones de los fármacos se miden en el plasma mediante punción venosa (de 2 a 5 mL de sangre). Sin embargo, el muestreo venoso es difícil en algunas poblaciones como los recién nacidos, los pacientes con flebitis, pacientes con tratamiento oncológico, en terapia intensiva y desnutridos, entre otros, ya que per se su condición no es favorable para la obtención de muestras sanguíneas con grandes volúmenes. En este sentido, la técnica de sangre seca ha venido a beneficiar enormemente el monitoreo farmacológico ya que los pacientes pueden ser monitoreados sin requerir grandes volúmenes de sangre y con menos invasión que con la técnica clásica de venopunción.18
La supervisión farmacológica sistemática sólo se aplica a un pequeño número de fármacos, principalmente aquellos con intervalo terapéutico estrecho como el metotrexato, la digoxina, el litio y los antiepilépticos, entre otros.25,26 La metformina no tiene un intervalo terapéutico estrecho pero es un fármaco que puede llegar a ser requerido a largo plazo o de por vida. Asimismo, comercialmente carece de una forma farmacéutica en concentración tal que pueda ser administrada en pacientes pediátricos. Por ello se corre el riesgo de no tener un efecto terapéutico o bien tener el efecto contrario (efecto tóxico). Diversos fármacos aprobados en adultos por la FDA no cuentan con la aprobación para su uso en pacientes pediátricos; no obstante, se empiezan a prescribir en niños que requieren instaurar el tratamiento farmacológico, por lo que es importante realizar estudios farmacocinéticos, farmacodinámicos, así como la supervisión de dichos medicamentos para establecer su eficacia y seguridad para la población infantil. En nuestro estudio se demuestra que el método analítico es adecuado para la cuantificación de metformina en muestras de pacientes pediátricos, así como en pacientes adultos. Existe un estudio previo utilizando la técnica de gota de sangre seca para vigilancia de metformina en sangre de pacientes con diabetes mellitus tipo II; sin embargo, en este ensayo se detecta el fármaco por UV a 236 nm, cuyo límite lineal es mayor al de este estudio (300-5000 ng/mL) y su límite de detección es de 150 ng/mL, mientras la detección por espectrometría de masas disminuye el límite a 50 ng/mL.27
Comportamiento farmacocinético de la formulación extemporánea
La formulación extemporánea líquida de metformina utilizada en este trabajo ha demostrado tener una farmacocinética similar a la de la tableta fraccionada en voluntarios humanos. Las diferencias encontradas, entre los parámetros farmacocinéticos de la formulación extemporánea y la tableta, es posible que se deban a que un fármaco disuelto se absorbe más rápidamente que uno sólido; de ahí que con la solución edulcorada se haya encontrado mayor concentración plasmática máxima, ya que accedió más rápidamente a su principal sitio de acción que es el hígado.
La sutil diferencia, de sólo 12 minutos en el Tmáx de la solución con respecto al de la tableta, sugiere que, efectivamente, la formulación extemporánea se absorbe más rápidamente que la tableta fraccionada. Este hecho era de esperarse pues toda presentación farmacéutica sólida administrada por vía oral exhibe un retraso en su absorción por el tiempo que tarda en fragmentarse en el sistema digestivo. La variación encontrada en la constante de eliminación sugiere que la metformina tardó menos en eliminarse cuando se administró previamente disuelta, aproximadamente una unidad menos que la tableta por cada dos minutos.
Del mismo modo, el perfil farmacocinético que se muestra en la Figura 1 sugiere que la eliminación de la metformina en forma disuelta fue sostenida y lineal, a diferencia de la tableta, que se eliminó de forma más abrupta. No obstante, las áreas bajo la curva (AUC0-12 y AUC∞) de ambas formas farmacéuticas tienen valores muy similares, es decir, que son hasta 50 veces más similares entre sí que los demás parámetros farmacocinéticos.
Lo anterior, sugiere que la biodisponibilidad por vía oral de la metformina, en la formulación extemporánea que hemos propuesto, es prácticamente la misma que la del medicamento en su forma original, sólo que ofrece la ventaja de tener dosis ajustadas más pequeñas que la tableta fraccionada y uniformidad de contenido.
CONCLUSIONES
Nuestra formulación extemporánea de metformina tiene biodisponibilidad comparable a la de la tableta fraccionada. Es igualmente biodisponible que la forma farmacéutica original, con la ventaja de permitir dosis menores que la tableta fraccionada, con precisión y uniformidad de contenido.

