











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las duplicaciones son rearreglos cromosómicos estructurales infrecuentes, en comparación con las deleciones o pérdidas de información genética. Las duplicaciones que involucran la región 5q35 se han asociado a un fenotipo inverso de síndrome de Sotos, por contener el gen NSD1 en triple dosis. Este síndrome se caracteriza por presentar talla baja, microcefalia, alteraciones en el aprendizaje, discapacidad intelectual leve/moderada, problemas de comportamiento y algunas dismorfias faciales como hipoplasia medio facial, fisuras palpebrales cortas con epicanto, nariz prominente, punta de la nariz hacia arriba, surco nasolabial liso, filtrum largo, boca pequeña, labio superior delgado, así como anomalías esqueléticas, craneosinostosis, hipoplasia de falanges, polidactilia preaxial, defectos cardiacos, criptorquidia y hernias inguinales.1,2 En cuanto a su origen, Dikow et al. describieron 4/14 (28.6%) casos de novo, 4/14 (28.6%) casos de origen materno y 6/14 (42.8%) se catalogaron como no determinados.1
En este trabajo se describe el caso de una paciente con diagnóstico de duplicación 5q34q35.3 de novo; se hizo una revisión de los genes involucrados en el fenotipo de la paciente. Es importante describir el caso para incluirlo dentro del grupo de pacientes con duplicación de esta región, con la delimitación precisa de la duplicación a nivel de nucleótidos, así como sus características fenotípicas, para de esta manera contribuir a la delineación del fenotipo de esta duplicación.
CASO
Niña de 3 años 8 meses de edad que cuenta con los siguientes antecedentes de importancia: producto de la gesta 5/5, madre de 38 años y padre de 42 años al nacimiento de la paciente, ambos sanos. Niegan consanguinidad y endogamia. Sin antecedentes heredofamiliares de importancia para el padecimiento actual. Embarazo normoevolutivo resuelto por vía abdominal a las 37 semanas de gestación, con peso 2,300 g, talla 47 cm y Apgar 9 a los 5 minutos.
En cuanto al desarrollo psicomotor, fijó mirada al 1 mes, sostén cefálico 2 meses, sonrisa social 1 mes, sedestación 1 año 3 meses, bipedestación con apoyo 1 año 2 meses, transferencia de objetos 1 año, inicia a vestirse y desvestirse 2 años 4 meses, brinca despegándose del suelo 2 años 6 meses, brincar con 1 pie 2 años 10 meses, sigue órdenes sencillas 2 años 3 meses (pasar objeto, cerrar puertas, apagar luces).
En el área del lenguaje: balbuceos 10 meses, monosílabos 1 año 3 meses, bisílabos 2 años. Actualmente: camina, corre, sube escaleras y baja, anda en bicicleta, come sola, se desviste sola, colorea, reconoce partes de su cuerpo, identifica algunas vocales y colores, obedece órdenes complejas (pasar objetos de un color en específico, llevar objetos a algún lugar en particular), acude a escuela regular con dificultades del aprendizaje, por lo que está pendiente su reingreso a escuela especial.
Al nacimiento se detectó alteración a nivel de cráneo, motivo por el cual es referida al Instituto Nacional de Pediatría. A su ingreso se interconsultó al servicio de Neurocirugía por el padecimiento de base, quienes a su vez, por las diferentes dismorfias faciales asociadas a las malformaciones presentes en la paciente, solicitan valoración del servicio de Genética, en donde a los 2 años 3 meses se observó paciente con peso de 10 kg (pc<5), talla 84 cm (pc<5), cráneo con plagiocefalia por antecedente de cráneo en trébol, asimetría facial, fisuras palpebrales almendradas, narinas permeables, mejillas prominentes, labio superior delgado, micrognatia; pabellones auriculares acopados, conducto auditivo externo estrecho; cuello corto; tórax normolíneo, ruidos cardiacos rítmicos, sin soplos; abdomen sin visceromegalias o hernias; extremidades íntegras, simétricas con arcos de movilidad completos y sin contracturas, tono normal, sin reflejos patológicos, sensibilidad conservada (Figura 1).
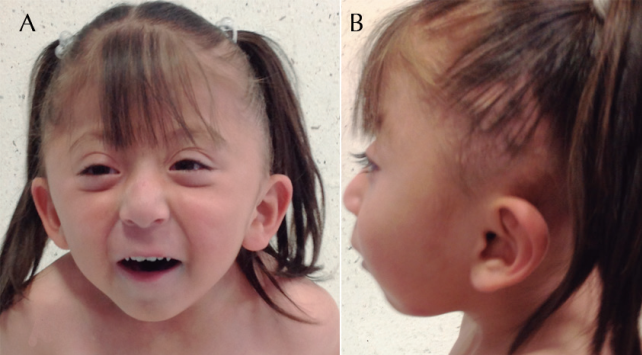
Figura 1 Fotografías de frente y lateral. A) Facies triangular con asimetría, fisuras palpebrales oblicuas hacia abajo y cortas, mejillas prominentes, labio superior delgado. B) Pabellones auriculares acopados con implantación baja, cuello corto.
Se inició abordaje como paciente dismorfológico con toma de sangre periférica para estudio citogenético, por presentar dos o más malformaciones mayores (craneosinostosis, cardiopatía congénita, peso y talla bajo y dismorfias faciales). El resultado del cariotipo mostró material adicional en el cromosoma 5, lo cual se corroboró posteriormente con FISH WCP.
La paciente presentó inicialmente alteraciones en la morfología craneal (cráneo en trébol), por lo que se solicitaron estudios de imagen (tomografía craneal) que confirmaron craneosinostosis de suturas múltiples. Por tal motivo, la paciente requirió de dos intervenciones quirúrgicas de remodelamiento craneal con avance frontotemporal, previo al año de vida. No presentó complicaciones operatorias ni postoperatorias. Actualmente, la paciente está asintomática, pero continua en seguimiento por el servicio de Neurocirugía.
Otra de las manifestaciones clínicas fue la presencia de soplo cardiaco; al ser valorada por el servicio de Cardiología se le encontró por ecocardiograma comunicación interventricular e interauricular que requirieron de corrección quirúrgica a los 4 meses de edad. No presentó complicaciones operatorias ni postoperatorias. Actualmente, la paciente continua en seguimiento por parte de este servicio.
En cuanto al desarrollo de la paciente, desde la primera valoración se detectó retraso del desarrollo psicomotor, para lo cual el servicio de Rehabilitación inició terapia física desde que contaba con 1 año 10 meses hasta los 4 años, momento en el que presentó recuperación completa de los hitos motores del desarrollo.
Por retraso en adquisición de lenguaje acudió al servicio de audiología donde por potenciales auditivos de tallo cerebral se descartó patología funcional, determinando audición normal bilateral y se recomendó terapia del lenguaje en algún centro de su lugar de origen. Sin embargo, perdió el seguimiento a los 3 años 6 meses. A la edad de 4 años 3 meses presentó problemas de aprendizaje, por lo que fue referida al servicio de Salud Mental donde mantiene seguimiento para valorar desarrollo de habilidades conductuales y de aprendizaje.
De igual forma, la paciente fue valorada por el servicio de gastronutrición por peso y talla bajos y se descartaron alteraciones gastrointestinales. A la edad de 5 años 2 meses de edad fue valorada por Endocrinología por la talla baja; determinaron patrón atenuado y velocidad de crecimiento bajo, con pruebas bioquímicas de la vía de los factores de crecimiento asociados a insulina (IGF, IGBP3) dentro de rangos normales y actualmente se mantiene en vigilancia.
Por último, la paciente cuenta con ultrasonograma renal que se reporta normal. Dicho estudio fue solicitado para descartar alteraciones asociadas a este nivel, que el padecimiento de base pudiera estar condicionando.
METODOLOGÍA
Inicialmente se realizó cariotipo con bandas GTG, de sangre periférica, como parte del estudio de pacientes con discapacidad intelectual y dismorfias mayores. Posteriormente, se realizó hibridación in situ con fluorescencia (FISH) en metafases de sangre periférica, con sonda WCP 5 (sonda para tinción de completa del cromosoma 5) de la marca Vysis ABBOTT.
A partir de una de muestra de ADN genómico (DNAg) obtenido de sangre periférica se realizó un microarreglo CytoScan HD de Affymetrix. Este tipo de microarreglo cuenta con 743,304 marcadores de SNP y 1,953,246 marcadores no polimórficos, con una resolución en genes de 25 marcadores por 100 Kb. A partir de 250 ng de ADN g se realizó la digestión con la enzima NspI incubando en un termociclador por 120 minutos a 37°C y 20 minutos a 65°C. Posteriormente, se ligaron los adaptadores NspI a los fragmentos utilizando la ADN ligasa T4 incubando 180 minutos a 16°C y 20 minutos a 70°C. Para la amplificación con reacción en cadena de la polimerasa se utilizaron los primers con la secuencia complementaria a los adaptadores y la enzima de alta procesividad Taq Titanium (Clontech), con el programa de amplificación: 30 s a 94°C, 45 s a 60°C y 15 s a 68°C, 30 ciclos; posteriormente, los productos se verificaron en gel de agarosa 2%; se purificaron con perlas magnéticas; se cuantificaron, fragmentaron y marcaron de acuerdo al protocolo del fabricante (Affymetrix). Finalmente, se realizó la hibridación por 18 horas a 50°C; después se tiñó y lavó el microarreglo, para concluir con el escaneo del mismo. El equipo que se utilizó fue el horno 645, la estación de fluidos 450 y el escáner 3000 7G de Affymetrix.
El análisis de las ganancias o pérdidas genómicas se realizó con el programa Chromosome Analysis Suite (ChAS) de Affymetrix, versión 2.0. El patrón de hibridación de la muestra nos permitió conocer el número de copias que se presentan a lo largo del genoma en diferentes regiones delimitadas por la posición de las sondas. El número normal de copias es de 2. Se considera que existe un cambio cuando el resultado del análisis de los microarreglos tiene un valor diferente de 2; será una ganancia cuando el número normal de copias tenga un valor de 3 o mayor y una pérdida cuando el valor sea de 1 o 0.
El contenido de esta región se analizó con ayuda de la página del UCSC Genome Browser (http://genome.ucsc.edu) y se comparó con la base de datos de las variantes genómicas (Database of Genomic Variants; http://projects.tcag.ca/variation) para excluir las variantes de cambios en el número de copias benignas. También se utilizaron las bases de datos del DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources) (https://decipher.sanger.ac.uk/), Clin Gen Dosage Sensitivity Map (http://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) y ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations) (http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp), como fuentes de información para la correlación genotipo-fenotipo.
RESULTADOS
El cariotipo reportó un cromosoma 5 con material adicional, 46,XX,add(5)(q35) en 15 metafases, con 400-500 bandas de resolución; el FISH con sonda para el cromosoma 5 reportó 46,XX,add(5)(q35).ish dup(5)(q35)(wcp5+) en 15 metafases, con lo cual se confirma que el origen del material adicional era del mismo cromosoma 5. El cariotipo con bandas G de ambos padres fue normal. Para definir puntos de ruptura se realizó microarreglo CytoScan HD de Affymetrix, el cual confirmó una duplicación intersticial de 17.6Mb (Figura 2),en la región q34q35.3 del cromosoma 5 (46,XX,add(5)(q35).arr[hg19] 5q34q35.3(163,110,984177,227,216x3,177,259,401179,330,764x3,179,346,465-180,719,789x3) dn) que contiene 138 genes (USCS genome browser; Figura 3).
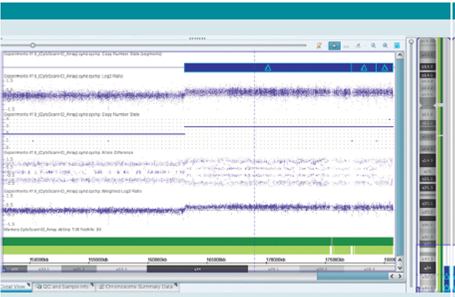
Figura 2 Microarreglo CytoScan HD de Affymetrix que muestra la duplicación de la región 5q35; 46,XX,add(5)(q35).arr[hg19] 5q34q35.3(163,110,984177,227,216x3,177,259,401179,330,764x3,179,346,465-180,719,789x3)dn.

Figura 3 Imagen obtenida de Genome Browser donde se muestran, señalados con flechas negras, los genes involucrados en el fenotipo de la paciente. El círculo verde indica el gen NSD1 asociado con talla baja y microcefalia; los cuadros rojos indican los genes MSX2 y FGFR4 asociados con craneosinostosis; y el círculo azul indica el gen NSX2-5 asociado con cardiopatía.
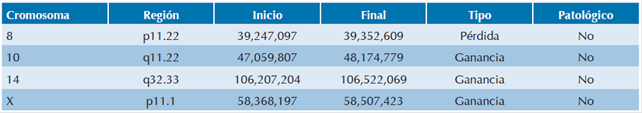
Además de este hallazgo, el análisis de los microarreglos de la paciente demostró 4 variantes en el número de copias (CNV) adicionales, distribuidas en 4 cromosomas diferentes (8, 10, 14 y X); 3 de ellas son con ganancia (CN=3) y solo 1 con pérdida (CN=1). De acuerdo con las bases de datos consultadas (Database of Genomic Variants, DECIPHER, Clin Gen Dosage Sensitivity Map y ECARUCA) las 4 variantes están descritas como no patológicas (Cuadro 1). La madre autorizó la toma de fotografías de la pacientes para lo cual firmó carta de consentimiento informado.
DISCUSIÓN
Los rearreglos cromosómicos estructurales a menudo son patológicos; sin embargo, existen algunos que se reportan como variantes normales, como la inversión pericéntrica del cromosoma 9. Se sabe que hasta el 75% de los rearreglos estructurales son de origen paterno, aunque también existen estudios que demuestran que tanto isocromosomas como duplicaciones invertidas son preferencialmente de origen materno.3 En particular, las duplicaciones se definen como la presencia de una copia extra de un segmento cromosómico que da origen a una trisomía parcial o completa, dependiendo de la porción del segmento cromosómico involucrado.
Los rearreglos cromosómicos pueden producir desequilibrios, ya sea pérdidas o ganancias de información genética, que pueden ocasionar algún fenotipo específico, y de acuerdo a lo descrito, el fenotipo de las ganancias o duplicaciones es menos grave que el fenotipo de un paciente que presenta deleciones.3 Cuando existe una duplicación cromosómica, el fenotipo va a depender de dos situaciones: por una parte, si el material adicional se inserta en tándem, es muy probable que las consecuencias clínicas sean sólo secundarias al exceso de copias de genes que se encuentran en el intervalo duplicado, pero si la adición del material se inserta en otro lugar del genoma, es posible que pueda provocar una disrupción de genes o elementos regulatorios, y como consecuencia el fenotipo se acompañe además de alteraciones provocadas por la disfunción génica del sitio de la inserción.4
El diagnóstico de estos rearreglos cromosómicos se puede realizar con métodos de citogenética convencional, en donde el cariotipo en prometafase nos permite detectar alteraciones cromosómicas estructurales de 5Mb, en comparación con el cariotipo que habitualmente se reporta de 550 bandas de resolución, que sólo logra detectar rearreglos mayores a 10Mb; el presente caso fue detectado por citogenética convencional debido a que la alteración tiene un tamaño mayor a 10Mb, frecuentemente establecido como el límite de resolución del cariotipo convencional, pero no permite delimitar con precisión la región involucrada e incluso tampoco permite definir el origen del material adicional, por lo cual es de suma importancia la aplicación de técnicas de mayor resolución para lograr dicho objetivo.
Con el avance de la tecnología la citogenética molecular y la citogenómica han logrado incrementar la detección de rearreglos complejos y menores a 3Mb. Entre estas técnicas están la hibridación in situ con fluorescencia o FISH (Fluorescence in situ hybridation) con sondas específicas, que se considera el ensayo de elección para localizar secuencias asociadas a un fenotipo característico;3,5 el FISH de tinción completa6 utilizado en la muestra de nuestra paciente fue útil para la visualización directa del cromosoma involucrado y a su vez confirmar que la duplicación es en tandem; y el estudio de microarreglos el cual nos permitió delimitar, con precisión a nivel de nucleótido, la región involucrada en la duplicación y por lo tanto los puntos de ruptura precisos a partir de los cuales se generó el rearreglo cromosómico.3,7
Una vez identificado el equilibrio cromosómico en un paciente, el siguiente paso es determinar si es un caso familiar o de novo, para lo cual se requiere el estudio de ambos padres del caso índice. En esta familia el estudio de cariotipo fue normal en ambos padres, por lo que se consideró como una alteración de novo, con un riesgo de recurrencia en la familia menor al 1%, aunque no se puede descartar la posibilidad de rearreglos crípticos balanceados en cuyo caso el riesgo de recurrencia se elevaría hasta un 50%.3
A lo largo del genoma existen regiones con alto contenido de repetidos o variaciones en el número de copias que son más susceptibles a recombinar entre sí, y que ahora se sabe están involucradas en la génesis de los rearreglos cromosómicos estructurales.3 Las duplicaciones generalmente son el resultado de un intercambio desigual entre cromosomas homólogos o cromátidas hermanas (Figura 4), que puede tener su origen en la meiosis de un portador de una translocación o por una inversión equilibrada.3 En nuestra paciente se demostró en el microarreglo un alelo extra, como se puede observar por las 4 líneas paralelas en la región de la duplicación, de lo que se desprende que el origen de este rearreglo fue un intercambio desigual entre cromosomas homólogos. (Figuras 2 y 4)
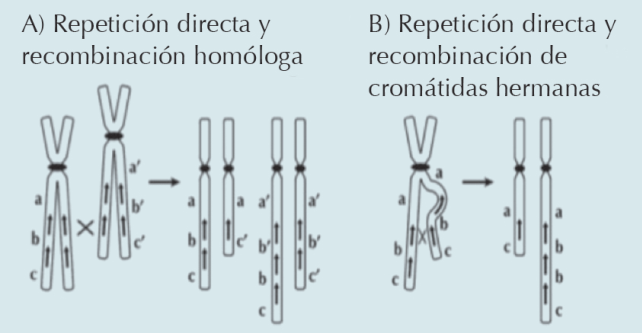
Figura 4 Los rearreglos cromosómicos se pueden producir por recombinación no alélica de homólogos entre secuencias parecidas o con repetidos con orientación idéntica (directos) u opuesto (inversos). A) Recombinación directa entre repetidos no alélicos en un cromosoma homologo. B) Cromátidas hermanas puede producir duplicaciones o deleciones complementarias. Tomado de Gersen S, Keagle M. 2013. Humana Press:139-174.
La duplicación de la región 5q34q35 se asocia con un fenotipo particular, que consiste en talla baja, microcefalia, alteraciones en el aprendizaje, discapacidad intelectual, problemas de comportamiento y algunas dismorfias faciales como fisuras palpebrales cortas, nariz prominente, punta de la nariz hacia arriba, surco nasolabial liso, labio superior delgado.1 En otras estadísticas se ha descrito también hipoplasia medio facial, pliegue epicanto, filtrum largo, boca pequeña, anomalía esqueléticas, craneosinostosis, hipoplasia de falanges, polidactilia preaxial, defectos cardiacos, criptorquidia y hernias inguinales2 (Cuadro 2). Cabe mencionar que el pronóstico de los pacientes con alteraciones cromosómicas estructurales dependerá de la región alterada, tipo de alteración y malformaciones asociadas. En el caso particular de la paciente, la adecuada evolución post quirúrgica y pocas complicaciones, así como la mejoría del desarrollo psicomotor, permiten establecer un pronóstico favorable a corto plazo, pero por la alteración cromosómica, la evolución de la paciente marcará el pronóstico a largo plazo.
Cuadro 2 Comparación de reportes previos en la literatura con nuestro paciente
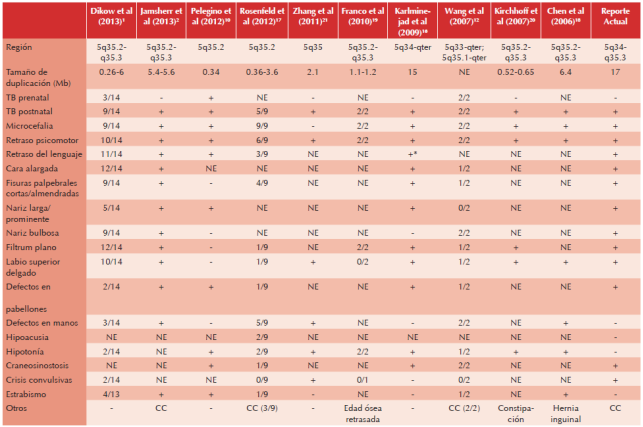
*Falleció a los 14 meses sin lenguaje, TB: talla baja; NE: no especificado; CC: cardiopatía congénita.
Dentro de esta región 5q34q35 existen varios genes que correlacionan con los datos clínicos presentes en los pacientes que ya han sido descritos y en esta paciente (Cuadro 3). De manera importante se encuentra el gen NSD1 (nuclear receptor-binding SET Domain-containingprotein-1) con locus en 5q35.3. Este gen codifica para un factor transcripcional nuclear; contiene 10 dominios conservados, uno de los cuales codifica para un dominio SET que contiene la función de metiltransferasa de histonas.8 La pérdida de función de este gen, incluyendo deleciones, es causante del síndrome de Sotos, cuyas manifestaciones son sobrecrecimiento, macrocefalia, dismorfias faciales y alteraciones en el aprendizaje.1,9 En contraste, las microduplicaciones que involucran esta misma región y por consiguiente la doble dosis del gen NSD1, se han asociado con talla baja y microcefalia, por lo cual un efecto de dosis en NSD1 podría ser la causa hipotética del también llamado fenotipo inverso de Sotos.1
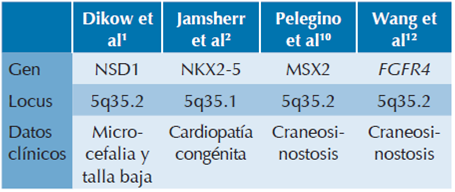
Otro gen localizado en esta región es el MSX2 (muscle segment homeobox-2), en el cual se ha demostrado que su sobreexpresión se asocia con craneosinostosis. Diversos estudios han demostrado que la elevación de dosis de MSX2 es suficiente para causar craneosinostosis, pero no otro tipo de malformaciones más severas.10-12 Además de MSX2, se sabe que el gen FGFR4 (fibroblast growth factor receptor 4) también se asocia a craneosinostosis, pero a diferencia de MSX2, FGFR4 también se relaciona con otras alteraciones óseas como agenesia radial y ausencia de pulgares.2,13 Los genes de la familia FGFR están involucrados en procesos celulares críticos incluyendo regulación del ciclo celular, migración, metabolismo, supervivencia, proliferación y diferenciación celular. Particularmente, el gen FGFR4 es un regulador importante de la osteogénesis con participación en la proliferación pre-osteoblástica, diferenciación y función osteoblástica durante la osificación intramembranosa.13
Otro gen importante en esta región es el gen NKX2-5, el que se ha relacionado con cardiopatía congénita en pacientes con duplicación de la región 5q34q35.2 Este gen es un factor de transcripción que regula la formación de los precursores de células miocárdicas en el desarrollo temprano del corazón. Las mutaciones en NKX2-5 se han asociado con varios defectos de septación, así como defectos en los conductos atrio-ventriculares.14 Algunas evidencias en modelos animales también sugieren que la sobreexpresión de NKX2-5 tiene un efecto adverso en el desarrollo cardiaco.15 Por lo tanto, el defecto cardiaco observado en estos pacientes pudiera explicarse por la sobreexpresión de NKX2-5.15 Otros estudios indican que no solamente la participación de NKX25 es responsable del fenotipo cardiaco de pacientes con duplicaciones parciales en 5q34, sino que también existen otros genes, entre ellos CSX1, así como elementos regulatorios localizados en la porción distal 5q que juegan un papel importante en el desarrollo de cardiopatía congénita.2
En el Cuadro 2 se hace una comparación entre los casos previamente descritos y lo encontrado en nuestra paciente. Como es de notarse, la extensión de la región duplicada va desde 0.26Mb hasta 15Mb.1,16 De acuerdo con nuestra revisión las características más frecuentes son: talla baja postnatal, retraso psicomotor, microcefalia, cara alargada, fisuras palpebrales cortas, nariz larga, filtrum plano, labio superior delgado, defectos en pabellones auriculares y craneosinostosis.17-21
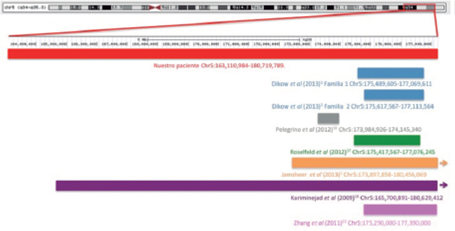
En la Figura 5 se muestra esquemáticamente los diferentes tamaños de duplicaciones de los casos previamente descritos y se hace una comparación con nuestra paciente. La duplicación de nuestra paciente es una de las más grandes informadas en la literatura (14Mb), siendo sólo superada por el reporte de Kariminejad et al16 de 15Mb. Es importante mencionar que nuestra paciente no tuvo manifestaciones clínicas extras en comparación con los casos publicados con duplicaciones más pequeñas, lo cual podría explicarse por la ausencia de genes causantes de patología en la región duplicada. Tampoco cursa con otros problemas mencionados en otros casos con la misma duplicación como malformaciones de manos, hipoacusia y estrabismo, que aunque no son tan frecuentes, su ausencia podría sugerir una expresividad variable.
CONCLUSIÓN
La incidencia de duplicaciones cromosómicas, como grupo heterogéneo de padecimientos genéticos, es de 1/4,000 recién nacidos vivos.22 Si bien no se cuenta con una incidencia de la duplicación de la región 5q34q35.3, consideramos de gran importancia la contribución de este reporte de caso para la delimitación del fenotipo de dicha entidad. Además, es importante mencionar que la técnica de microarreglos es indispensable para la búsqueda de la alteración precisa en pacientes dismorfológicos con cariotipo no concluyente e incluso es necesario para una adecuada correlación genotipo-fenotipo y, como consecuencia de esto, otorgar un asesoramiento genético lo más certero posible.

