











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink¿Cómo sabemos dónde están los átomos? Necesitamos la interacción con la materia en la minúscula escala atómica: sólo cosas del tamaño de los átomos pueden verlos. Pero los átomos son muy pequeños, del tamaño de una diezmilmillonésima de metro o un Angstrom. ¿Qué interacciones pueden emplearse?
El fenómeno de difracción permitió asomarnos a esa escala: irradiemos la materia con radiación electromagnética del tamaño de los átomos y esa radiación será reflejada; aseguremos que haya muchos átomos orientados de la misma manera ‒tantos como hay en los cristales periódicos‒ y, para el ángulo de reflexión correcto, la interacción constructiva entre la radiación proporcionará una relación de señal/ruido suficientemente notable para deducir la distancia entre los átomos ordenados periódicamente en esos planos. El laborioso trabajo de acumular y analizar numerosos planos permite deducir la estructura de rayos X ‒la posición espacial‒ de los átomos que constituyen el cristal. Premio Nobel a los Bragg en 1915 por los fundamentos, Nobel a Watson y Crick en 1962 por su aplicación exitosa en la deducción de la estructura del ADN, Nobel a Hodgkin en 1964 por su aplicación exitosa en la deducción de estructuras de proteínas y Nobel a Shechtman en 2011 por la inesperada existencia de cuasicristales (Amador Bedolla, 2012). Los rayos X interactúan con los minúsculos átomos y nos revelan su acomodo espacial. Sólo aplica a cristales periódicamente ordenados y se requiere un análisis numérico profundo de los resultados experimentales ‒que se puede hacer a mano pero que es mucho más rápido si se hace en computadora.
El fenómeno de resonancia permitió asomarnos a esa escala: busquemos la frecuencia de variación de un campo magnético que permita la modificación del valor del momento angular de espín de un núcleo. La relación señal/ruido en el fenómeno de resonancia es inmensa; la modificación del valor de la frecuencia necesaria para la resonancia es finísima: muy pequeña y de muy precisa determinación, además depende del ambiente químico en el que se encuentra el núcleo. La resonancia magnética nuclear permite deducir qué átomo está junto a qué otro y en la cercanía de cuál más. Premio Nobel a Bloch y Purcell en 1952 por los fundamentos, Nobel a Ernst en 1991 por el incremento en la resolución y la sensibilidad de la técnica, Nobel a Wüthrich en 2002 por su aplicación exitosa al análisis de proteínas, Nobel a Mansfield y Lauterbur en 2003 por su aplicación exitosa a la obtención de imágenes detalladas, in vivo, de órganos a cuál más vital (Boesch C., 2004). Las matemáticas y la computación que se requiere para desintricar estos datos exigió trabajo muy serio y en la actualidad disfruta de las ventajas de las computadoras más modernas.
El Nobel de Química de 2017 premia otro método que nos permite asomarnos a la escala atómica y deducir la posición de los átomos (Stokstad E., Callaway E., 2017). Este método se ha desarrollado a lo largo de más de cuarenta años y es particularmente útil para tratar estructuras biológicas por lo que constituye la mejor manera de deducir la compleja geometría de las proteínas; se trata de la microscopía crioelectrónica de una sola partícula (cryo-Electron Microscopy o cryo-EM) (Nogales E., Eisenstein M., 2016). Vayamos por partes.
Los electrones acelerados pueden interactuar también en la minúscula escala atómica, a condición de que los aceleremos suficientemente para que su dualidad onda-partícula ajuste el tamaño requerido. El plan sería entonces acelerar electrones, apuntarlos a que choquen con la muestra que se quiere determinar y medir para dónde salen reflejados/dispersados/difractados. Pero desde ahí empiezan los problemas, porque los electrones ‒con una carga eléctrica inmensa‒ interactúan con todo, todo los detiene, todo alteran: el medio por donde viajan hacia el objetivo ‒o sea que hay que hacerlos viajar en alto vacío‒, el medio en el que esté el objetivo ‒por ejemplo, el solvente en el que se encuentra la proteína y que opaca la distinción entre los átomos del solvente y los de la molécula que se quiere ver‒, la profundidad, dentro de la muestra, que pueden alcanzar los electrones ‒al interactuar con todo son detenidos en la superficie de la muestra‒ y el efecto destructivo de esos proyectiles sobre la muestra ‒el golpeteo de muchos electrones acelerados acaba por destruir la proteína. Hace cuarenta años no se veía fácil la solución de estos problemas.
Quizá sea justo decir que tres avances fundamentales permitieron que, a lo largo de esos cuarenta años, se desarrollara la técnica hasta el nivel presente que justificó la asignación del Premio Nobel reciente ‒un avance fundamental por cada uno de los tres premiados.
Jacques Dubochet, Universidad de Lausanne
Primero hay que preparar la muestra para que los electrones choquen con ella, reboten y sean detectados. Desde los años ochenta se propuso la idea que ha evolucionado hasta el presente. Colóquese una capa de carbono altamente poroso ‒una malla porosa con muchos huecos de radio micrométrico‒ sobre una placa metálica que sirve sólo de soporte. Aplíquese la solución de proteínas sobre la malla cuidando que el líquido penetre en los huecos. Límpiese la solución sobrante de la superficie de la malla, aquella que no penetró en los huecos. Congélese la solución a temperatura de criogenia (para el solvente usado típicamente, etano, esto es por debajo de
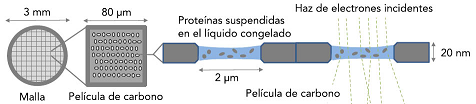
Los electrones van a salir en todas direcciones. Originalmente se usó una película fotográfica para detectar los electrones que rebotan, pero hay que considerar que la película fotográfica detecta en promedio sólo la mitad de los electrones. Algunos de los electrones rebotados provienen de la interacción con el objetivo ‒la proteína‒, otros provienen de la interacción con el solvente ‒que, al ser enfriado a gran velocidad no condensa en forma cristalina sino vítrea‒; para analizar los resultados hay que encontrar la forma de distinguir cuáles vienen de dónde, esta relación señal/ruido no es muy grande y se requerirán muchas mediciones para permitir la selección adecuada. Los electrones que se logran detectar en la película fotográfica forman una imagen bidimensional de la que se pretende deducir la estructura tridimensional de la proteína. La penosa comparación de las señales provenientes de cada una de las proteínas ‒cada una de ellas orientadas particularmente‒ permite deducir la información tridimensional a partir de múltiples señales bidimensionales cada una de ellas correspondiente a una proteína individual. De aquí el nombre de microscopía crioelectrónica de una sola partícula ‒aunque para acumular la información se requiera la medición de hasta cientos de miles de proteínas.
Joachim Frank, Universidad de Columbia
Múltiples partículas sencillas, orientadas de distinta manera, reciben el bombardeo de electrones enviados en la misma dirección. Las señales provenientes de la misma región de la proteína deben ser similares, una vez tomada en cuenta su diferente orientación. La combinación de las respuestas de miles de partículas sencillas acumulará evidencia de cómo es la estructura de cada una de las regiones. Este trabajo es impráctico para realizarse humanamente y requiere del empleo de computadoras. El avance extraordinario del hardware computacional durante los últimos cuarenta años ayudó a la microscopía crioelectrónica, como a tantos otros campos científicos; pero, al igual que ellos, ésta requirió también de avances específicos en el software.
Matemáticas aplicadas del más alto nivel. Porque, como ya se mencionó, los problemas son múltiples: los electrones son dispersados tanto por la proteína como por el solvente vítreo ‒solo 30% más por la proteína‒, pero no se pueden enviar muchos electrones porque éstos rompen enlaces y destruyen la proteína. Hay que distinguir la orientación de cada una de las partículas simples para las que se acumulan datos escasos y borrosos y combinar la información bidimensional para deducir la información tridimensional. Las técnicas matemáticas estadísticas empleadas reciben nombres como análisis de componentes principales (Carroni M., Saibil H. R., 2016), métodos de máxima probabilidad (maximum-likelihood), el abordaje empírico de la estadística Bayesiana (Fernandez-Leiro, R., Scheres S. H. W., 2016), métodos de ajuste flexible de dinámica molecular y su culminación reciente en el código RELION (Frank J., 2016).
Richard Henderson, MRC Laboratory Cambridge
Henderson intentó por primera vez el empleo de la microscopía electrónica para estudiar la estructura de proteínas en la década de los años setenta del siglo pasado. Su participación añade a esta contribución seminal la responsabilidad de resolver el otro problema central de la técnica treinta años después. La recolección de los datos que indican a dónde fueron desviados los electrones se hizo inicialmente con película fotográfica. A principios de este siglo Henderson promovió su remplazo por detectores electrónicos digitales, conocidos como aparatos digitales de carga acoplada (digital charge-coupled device) (Callaway E., 2017). Estos aparatos no sólo han permitido el aumento de la resolución de la señal sino que permiten acumularla a mucha mayor velocidad que la película fotográfica, y automatizar su detección.
La mayor resolución de la detección digital ha permitido una aplicación inesperada: las distintas partículas corresponden a proteínas que fueron capturadas ‒antes de la criogenia, cuando estaban a temperatura ambiente‒ en distintos estados de su movimiento característico. La detección digital acumula tanta información que se estudia ahora la diferencia entre esas estructuras para detectar cómo actúan éstas in vivo.
Multidisciplina
El premio Nobel de Química 2017 encierra una lección particular. La aplicación presente de la crio-microscopía electrónica de una sola partícula la define como la fuente más eficaz para conocer la estructura de moléculas bioquímicas, particularmente de proteínas, para las que destaca dos habilidades casi imposibles para las técnicas alternativas: la detección de estructuras de proteínas de membrana y la detección de su dinámica funcional. Estas capacidades justifican ampliamente la asignación del premio: la bioquímica ha avanzado y avanzará considerablemente gracias a esta técnica.
Sin embargo, los avances que permitieron el desarrollo de la técnica, y que fueron llevados a cabo por investigadores del área bioquímica, pertenecen a disciplinas distintas. Ya vimos que la posibilidad de emplear la microscopía electrónica ‒que se puede asignar al campo de la física‒ requirió los detalles técnicos de la criogenia y el uso de carbono poroso ‒¿quizá química?‒; que la acumulación de información procedente de miles de moléculas y la deducción de estructuras en 3D a partir de datos de 2D requirió de la estadística matemática y el software más avanzado y que la detección eficiente de los electrones dispersados requirió del empleo de aparatos de la electrónica de última generación. Física, química, matemática, ciencias de la computación y electrónica para hacer avanzar la bioquímica. Así es la investigación moderna.
Aplicaciones y futuro
Cerca de 90% de las estructuras de proteínas conocidas se han obtenido mediante el uso de cristalografía de Rayos X. Las estructuras del 10% restante se han obtenido mayoritariamente empleando RMN y menos de 1% del total a la fecha proviene de la crio-microscopia electrónica. Sin embargo, esta novedosa técnica es particularmente útil para encontrar estructuras de proteínas presentes en la membrana celular que son casi imposibles de obtener por las otras técnicas. La joya de la corona la constituye la muy reciente elucidación de la proteína
El futuro de la microscopía crioelectrónica de una sola partícula es promisorio. Los obstáculos que se han podido resolver en los cuarenta años que han transcurrido desde la concepción de la posibilidad de determinar estructuras de proteínas por irradiación de electrones hasta el presente son extraordinarios. Y su distinción con el premio Nobel es totalmente merecida. Pero las expectativas de futuro que esta metodología plantea son igualmente sorprendentes. Destacan dos aspectos que se extienden, en la actualidad, a muchos otros campos científicos (Aspuru-Guzik et al. 2018): la automatización (robotización) de las técnicas de laboratorio necesarias para aplicar el principio y el empleo de inteligencia artificial/aprendizaje de máquina para optimizar el análisis de la estructura a partir de los datos detectados.

