











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La contaminación de las aguas superficiales y subterráneas debido a las descargas coloreadas emitidas por las industrias textiles, cosméticas, farmacéuticas, del cuero, del papel y alimenticias generan graves problemas ambientales (Wang et al. 2014), ya a que un 15 % de los colorantes se pierden durante los procesos de producción y aplicación (Wang et al. 2008, Jagruti 2015). Los colorantes azoicos contribuyen con aproximadamente el 70 % de dichos desechos (Matouq et al. 2014) debido a la facilidad de los procesos de obtención, sus elevados coeficientes de extinción molar y su media a alta estabilidad frente a la luz, la humedad y los ataques microbianos (Bafana et al. 2011).
La toxicidad aguda de los colorantes azoicos es relativamente baja, dado que presentan valores de dosis letal para el 50 % de la población (DL50) en el rango de 250-2000 mg/kg de peso corporal (Bafana et al. 2011). Algunos colorantes azoicos (los clasificados como ácidos, básicos y directos) son tóxicos agudos para peces, crustáceos, algas y bacterias, mientras que los colorantes azoicos reactivos poseen valores de concentraciones efectivas muy altas (CE > 100 mg/L), por lo que son considerados no tóxicos para los organismos acuáticos (Novotny et al. 2006). La acción carcinogénica y/o mutagénica de los colorantes azo puede deberse a la acción del compuesto como tal, a la formación de arilaminas a través de los procesos reductivos durante las biotransformaciones (Chung et al. 1992, Collier et al. 1993, Rajaguru et al. 1999) o a los productos obtenidos a través de la vía oxidativa del sistema citocromo P450 (Arlt 2002, Umbuzeiro et al. 2005, Wang et al. 2012). Por estas razones, la eliminación del agua de estos compuestos es muy importante para la seguridad del ambiente y la salud humana (Wu et al. 2012).
Las técnicas propuestas para tratar los efluentes que contienen colorantes son numerosas, encontrándose entre ellas la coagulación y la floculación (Sanghi et al. 2007, Fang et al. 2010, Moghaddam et al. 2010), la electrocoagulación (Essadki et al. 2008, Eyvaz et al. 2009), la adsorción sobre diferentes materiales (Crini 2006, Demirbas 2009, Royer et al. 2009, Ahmad y Hameed 2010, Mui et al. 2010), el intercambio iónico (Karcher et al. 2002, Greluk y Hubicki 2013), el tratamiento con ozono (Shu y Huang 1995, Peralta-Zamora et al. 1999, Wu et al. 2008, Tehrani-Bagha et al. 2010), la fotocatálisis (Konstantinou y Albanis 2004, Guettai y Amar 2005, Rauf y Ashraf 2009, Ahmed et al. 2011) y la biodegradación, ya sea mediante hongos (Paszczynski et al. 1992, Robinson y Nigam 2008, Kaushik y Malik 2009, Karthikeyan et al. 2010, Levin et al. 2010), algas (Omar 2008, Lim et al. 2010, Grassi et al. 2011) o bacterias (Khehra et al. 2005, Joshi et al. 2010, Modi et al. 2010, Phugare et al. 2011, Saratale et al. 2011). Los procesos de oxidación avanzados (POA) son métodos fisicoquímicos basados en la producción y utilización de radicales HO para tratar efluentes, y se ha demostrado que son una herramienta efectiva para aguas coloreadas (Wang 2008, Arslan-Alaton et al. 2009, Mahamuni y Adewuyi 2010, Kalra et al. 2011, Dutta et al. 2015, Guo et al. 2015, Wang et al. 2017).
Entre los POA, el tratamiento mediante ultrasonido se presenta como una metodología atractiva debido a sus ventajas. Los efectos químicos del ultrasonido se deben al fenómeno de cavitación acústica (Adewuyi 2005), el cual involucra la formación, crecimiento y colapso de microburbujas, lo que da lugar a la existencia de “puntos calientes” con condiciones extremas de temperaturas y presiones no alcanzables por métodos tradicionales, superiores a los 5000 K y 1000 atm (Gogate 2008, Eren e Ince 2010, Eren 2012, Kieffer et al. 2015, Yuan et al. 2016). En estas condiciones extremas se obtiene una gran cantidad de especies reactivas (Pang et al. 2011):
Existe una gran cantidad de trabajos (Wang et al. 2007, Bejarano-Pérez y Suárez-Herrera 2008, Okitsu et al. 2008, Ghodbane y Hamdaoui 2009, Guo et al. 2010) donde se reporta el uso de tetracloruro de carbono como agente sinérgico, ya que este compuesto disminuye la recombinación de radicales oxhidrilos (ecuación 11), sustrayendo H• y dando lugar a la formación de nuevas especies altamente reactivas:
Para el TM (que también es secuestrante de radicales hidrógenos), además de las ecuaciones 13 a 17, pueden agregarse las siguientes reacciones:
Bhatnagar y Cheung (1994) señalan que la degradación sonoquímica del TM en medio acuoso sigue una cinética de falso primer orden, con un valor de la constante de velocidad de 0.043 1/min, siendo el HCl el único producto detectado.
En este estudio se eligió al anaranjado de metilo (ácido 4-(((4-dimetilamino)fenil)azo)benzeno sulfónico), un colorante azoico utilizado en la industria textil que puede causar irritación al contacto con la piel, los ojos, el sistema respiratorio superior, y que puede ocasionar irritaciones, náusea, vómito y diarrea si es ingerido (TTI 2016). Si bien puede clasificarse como un compuesto moderadamente tóxico en caso de una ingestión única DL50 = 60 mg/kg (oral, ratas) y 101 mg/kg (intraperitoneal, ratones) es un colorante considerado mutagénico de acuerdo con ensayos realizados en células humanas (fibroblastos), en células embrionarias en ratones y en células del riñón en hams-ters (Sigma-Aldrich 2004). Por estas razones, el AM es muy utilizado como colorante modelo en estudios de degradación de contaminantes (Marci et al. 2003, Zhang et al. 2006, Ali y Abdullah 2011, Haji et al. 2011, Kodom et al. 2012, Urmi et al. 2015, Tang 2013, Hakamada et al. 2014, Zhao et al. 2014, Wang et al. 2015, Zyoud et al. 2015, Youssef et al. 2016, Yuan et al. 2016). Nuestro trabajo también lo hace, planteando como objetivo la posibilidad de utilizar el ultrasonido como tratamiento para aguas coloreadas con AM, analizando el efecto del uso de triclorometano sobre la velocidad de decoloración, y utilizando un reactor por lotes y uno continuo. Se eligió al TM como alternativa al tetracloruro de carbono, por ser el primero más soluble en agua y poseer mayor presión de vapor, lo que favorece su ingreso en las burbujas de cavitación.
Guivarch et al. (2003) propusieron un mecanismo de reacción para la degradación completa del AM (hasta CO2 y H2O), cuyas primeras reacciones se muestran en la ecuación 23. Dado que en nuestro trabajo no se analizaron los compuestos intermedios, no se pudo corroborar si los productos de degradación son los mismos durante la decoloración sonoquímica.
donde NO2 Ph = nitrobenceno, NO2 Ph OH = p-nitrofenol y HO Ph OH = 1,4 - dihidroxibenceno.
MATERIALES Y MÉTODOS
Reactivos y equipos analíticos
Se utilizó AM grado analítico Mallinckrodt (PM: 327.34 g/mol, CAS: 547-58-0) y triclorometano proanálisis Cicarelli (PM: 119.38 g/mol, densidad 1.49 g/mL, CAS: 67-66-3). Para preparar las soluciones se utilizó agua ultrapura (osmosis inversa, desionizada) de conductividad < 0.05 mS/cm. Las concentraciones de AM se midieron por espectrometría visible en un equipo HATCH DR/2010 a 465 nm (Cheng et al. 2012). Las determinaciones de pH se efectuaron con un equipo HATCH Sension PH1. Todos los ensayos de decoloración (tanto en el reactor por lotes como en el continuo) se hicieron por triplicado.
En el reactor por lotes el valor medio de la concentración inicial de las soluciones de AM fue de 3.45 mg/L ± 0.04 mg/L para los ensayos sin agregado de TM, mientras que para los ensayos con TM fueron de 3.88 mg/L ± 0.02 mg/L. En ambos casos el valor del pH inicial fue de 5.7. Las concentraciones iniciales de AM y los valores de caudales utilizados en el reactor continuo se muestran en el cuadro I.
CUADRO I PROMEDIOS Y DESVIACIONES ESTÁNDAR DE LAS CONCENTRACIONES Y CAUDALES UTILIZADOS EN EL REACTOR CONTINUO
| Concentraciones (promedio/desviación estándar) (mg/L) | Caudales (promedio/desviación estándar) (L/h) | |
| Con 1 mL de HCCl3/L | 3.55/0.11 | 9.8/0.3 |
| 5.10/0.10 | 9.5/0.2 | |
| 6.95/0.20 | 9.2/0.3 | |
| 8.96/0.37 | 10.2/0.5 | |
| Sin HCCl3 | 1.97/0.10 | 9.7/0.1 |
Equipos de ultrasonido
El reactor sonoquímico por lotes es un equipo MSE provisto de un cabezal de 13 mm de diámetro, que opera a una frecuencia de 20 kHz sumergido 2 cm en el líquido. La potencia entregada por el equipo fue determinada mediante calorimetría (Thompson y Doraiswamy 1999) en experiencias efectuadas por triplicado, que dieron como resultado una potencia efectiva de 35.3 W ± 0.58 W. Se utilizó un reactor de vidrio con capacidad de 250 mL, con camisa de refrigeración, por la que se hizo circular agua para regular la temperatura de operación a 25 ± 1 ºC.
El reactor continuo (construido por el Grupo de Química Ambiental, Fig. 1) está compuesto por cuatro unidades: la primera de 0.530 L (20 kHz y 33.6 W ± 0.79 W), la segunda de 0.526 L (40 KHz y 17 W ± 0.75 W), la tercera de 0.529 L (20 kHz y 27.0 W ± 0.42 W) y la cuarta de 0.535 L (40 kHz y 49.7 W ± 0.23 W). Las cuatro unidades conforman un solo cuerpo y cada una descarga en la siguiente a través de un rebosadero. La alimentación del agua a tratar se efectúa mediante una bomba peristáltica. Cada unidad tiene un sistema de ventilación forzada para mantener temperaturas estables (en estos ensayos las mismas variaron entre 24 y 28 ºC).
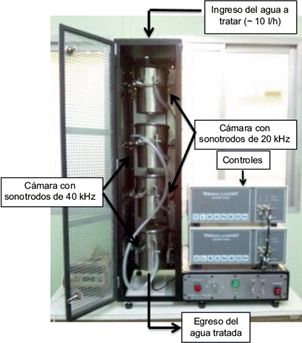
Programas utilizados
Los datos se analizaron mediante la hoja de cálculo Gnumeric (2014). Se realizó un modelo del funcionamiento del reactor continuo mediante el lenguaje simbólico XCOS (Scilab 2017) con el fin de verificar el tiempo de residencia, utilizando para cada una de las unidades el balance de masas indicado en la siguiente ecuación:
donde Q = caudal, i = número de la unidad (i = 1 - 4), C i = concentración en la unidad i (C 0 = concentración de ingreso), V i = volumen de la unidad i y K i = constante cinética de primer orden.
RESULTADOS
Reactor por lotes
AM sin agregado de HCCl 3
En la figura 2 se muestra la evolución de las concentraciones (expresadas en porcentajes de la concentración inicial) en función del tiempo, así como las variaciones de pH. Las barras verticales indican el rango entre los valores máximo y mínimo, mientras que los círculos el valor medio de las tres medidas. Para el caso del pH se grafican únicamente los valores medios. Se muestran además las ecuaciones de ajuste (de seudoprimer orden en el caso del AM y polinómica de segundo orden para el pH) con los respectivos coeficientes de determinación.

AM con agregado de HCCl 3
En la figura 3 se presentan los resultados de los ensayos realizados con el agregado de tres concentraciones diferentes de cloroformo (1, 2 y 3 mL/L) (se grafican únicamente los valores medios). En el cuadro II se muestran los valores de las constantes cinéticas de decoloración de primer orden y de seudoprimer orden ajustadas a los datos mostrados en la figura 3, junto con los respectivos coeficientes de determinación, que en todos los casos son estadísticamente significativos. Los resultados indican un hecho ya observado por otros autores para el CCl4 (Bejarano-Pérez y Suárez-Herrera 2008, Chakinala et al. 2008, Guo et al. 2010): que la velocidad aumenta a medida que se incrementa la concentración de HCCl3 hasta un máximo a partir del cual comienza a decaer lentamente. En nuestro caso, dicho máximo se presentó para el agregado de 2 mL/L.

Fig. 3 Decoloración del anaranjado de metilo con diferentes concentraciones de HCCl3 en el reactor por lotes
Comparación de resultados
Con el fin de analizar el efecto sinérgico (ES) Madhavan et al. (2010) y Cheng et al. (2012) propusieron utilizar relaciones entre las constantes de velocidad. En nuestro caso éstas quedan expresadas en la siguiente ecuación:
Utilizando los datos del cuadro II (el valor de la constante cinética de primer orden igual a 0.0042 1/min para la decoloración sin triclorometano y la no decoloración del AM con HCCl3) se obtienen los datos mostrados en el cuadro III.
CUADRO III EFECTO SINÉRGICO (ES) DEL AGREGADO DE TRICLOROMETANO EN EL REACTOR POR LOTES
| mL HCCl3/L solución | 1 | 2 | 3 |
| ES | 421.4 | 632.9 | 560.0 |
Para verificar estadísticamente el efecto sinérgico se compararon las constantes de primer orden sin TM y la correspondiente al agregado de 1 mL/L de TM (con la cual se observa el menor porcentaje de decoloración). En el cuadro IV se presentan los datos del cálculo de la homocedasticidad utilizando el estadístico F de Snedecor, así como los correspondientes a la prueba t de Student para varianzas inhomogéneas. Los resultados muestran que ambas constantes son estadísticamente diferentes (con un nivel de confianza del 95 %).
CUADRO IV ANÁLISIS DE LA HOMOCEDASTICIDAD DE LAS VARIANZAS Y DE LAS DIFERENCIAS ENTRE LAS CONSTANTES DE DECOLORACIÓN
| Sin el agregado de TM | Con el agregado de TM | |
| Constante de primer orden | 3.99 10-3 (1/min) | 1.740 (1/min) |
| Varianza de la constante | 4.667 10-8 | 2.141 10-2 |
| Grados de libertad | 5 | 9 |
| F calculado | 5.59 105 | |
| F tabulado (95 %) | 4.772 | |
| t calculado | 11.863 | |
| t tabulado (95 %) | 2.262 | |
TM: triclorometano, F: estadístico de Snedecor, t: estadístico de Student
Reactor continuo
En la figura 4 se muestra el porcentaje de decoloración del AM para una concentración baja sin el agregado de cloroformo y para otras cuatro concentraciones mayores con el agregado de 1 mL/L de TM.

Fig. 4 Decoloración del anaranjado de metilo en reactor continuo. A = 3.55 mg/L, B = 5.10 mg/L, C = 6.85 mg/L, D = 8.96 mg/L, E = 1.97 mg/L. A-D: con el agregado de 1 mL/L de HCCl3; E = sin el agregado de HCCl3
Se observa que cuando el equipo entra en estado estacionario sin el agregado de HCCl3, se obtiene un porcentaje de decoloración inferior al 10 %, mientras que con el agregado (para las cuatro concentraciones analizadas) se obtiene un porcentaje superior al 80 %.
Para calcular el efecto sinérgico se consideraron los porcentajes de decoloración (Pd) en estado estacionario, con corrección por las concentraciones de AM (CAM) (dado que las utilizadas en los ensayos sin y con cloroformo fueron diferentes), es decir:
Los resultados obtenidos mediante la aplicación de la ecuación 26 se muestran en el cuadro V. No son comparables con los obtenidos en el reactor por lotes debido a la diferencia en las ecuaciones de cálculo.
Simulación dinámica
Utilizando el editor gráfico XCOS se realizó un modelo de simulación dinámica del reactor continuo. Éste se presenta en la figura 5 a, b, donde se muestran, respectivamente, las cuatro unidades con sus ingresos y egresos, así como un detalle de la primera unidad (los paneles restantes son similares).

Fig. 5 Superior: diagrama de bloques del reactor continuo. Inferior: detalle del diagrama de bloques correspondiente a la unidad
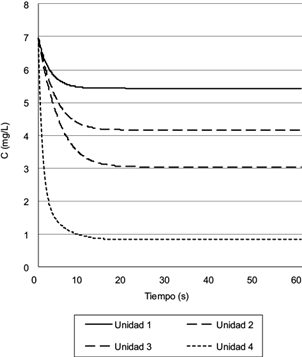
En la figura 6 se muestra la salida de la simulación dinámica para cada una de las cuatro unidades del equipo continuo, utilizando como concentración de entrada 6.94 mg/L de AM y un caudal de 9.4 L/h (correspondiente a uno de los ensayos realizados), así como el agregado de 1 mL/L de HCCl3.
DISCUSION
Se demostró que la decoloración sonoquímica de AM es ampliamente favorecida por la adición de bajas concentraciones de HCCl3. En el caso del reactor por lotes se obtuvieron degradaciones del 17 y el 90 % sin y con este agregado. Este aumento en la velocidad puede explicarse por el hecho de que el cloroformo es más volátil que el agua (presiones de vapor: pvagua(27 ºC) = 26.74 Torr; pvcloroformo (30 ºC) = 160 Torr) lo que favorece su ingreso en las burbujas de cavitación; al colapsar éstas dan lugar a las reacciones expresadas en las ecuaciones 19-21, secuestrando radicales H•, lo cual permite un mayor tiempo de vida medio de los radicales HO• y, por ende, mayor probabilidad de reaccionar. También puede ocurrir la reacción expresada en la ecuación 22, pero debido a que los enlaces C-H tienen mayor fuerza que los enlaces C-Cl, se produce en menor extensión. Para las tres concentraciones de HCCl3 empleadas el efecto sinérgico es superior al 400 %.
En el cuadro VI se presenta una recopilación de métodos utilizados para la degradación de AM en reactores por lotes, incluyendo los datos de este trabajo. Más allá de las diferencias observadas en los valores de frecuencias y potencias de los tratamientos utilizando únicamente ultrasonido, en un solo trabajo se obtiene una degradación del 52 %, mientras que en los restantes nueve casos (incluido el nuestro), los porcentajes de degradación son inferiores al 20 %: Esto muestra que el método es inadecuado para este fin.
CUADRO VI DEGRADACIÓN DEL ANARANJADO DE METILO (AM) EN REACTORES POR LOTES MEDIANTE DIFERENTES PROCESOS
| Concentración inicial AM | Proceso | Degradación (%) | Tiempo | Constante de degradación | Referencia |
| 100 mg/L | US (40 kHz - 400 W) | 0 | 120 min | --- | Chen et al. 2008 |
| Fenton | 80 | ||||
| US (40 kHz - 400 W) + Fenton | 86 | ||||
| 10 ppm | UV + ZnO | 100 | 60 min | --- | Zyoud et al. 2015 |
| 40 ppm | UV + ZnO | 40 | |||
| --- | US (400 W) | 5 | 90 min | --- | Cheng et al. 2012 |
| TiO2 (2 g/L) | 8 | --- | |||
| US (400W) + TiO2 (2 g/L) | 13 | 0.0025 1/min | |||
| UV (11 W) + TiO2 (2 g/L) | 66 | 0.0125 1/min | |||
| US (400 W) + UV (11 W) + TiO2 (2 g/L) | 93 | 0.0310 1/min | |||
| 25 mg/l | US (28 kHz - 400 W) - pH: 7 | 15 | 100 min | 0.00155 1/min | Cui et al. 2011 |
| TiO2 (0.5 g/L) + UV (30 W) - pH: 7 | 28 | 0.00323 1/min | |||
| US (28 kHz - 400 W) + UV (30 W) + TiO2 (2 g/L) - pH: 7 | 33 | 0.00388 1/min | |||
| US (28 kHz - 400 W) - pH: 4 | 15 | 0.00155 1/min | |||
| TiO2 (0.5 g/L) + UV (30 W) - pH: 4 | 41 | 0.00500 1/min | |||
| US (28 kHz - 400 W) + UV (30 W) + TiO2 (2 g/L) - pH: 4 | 62 | 0.00938 1/min | |||
| 10 ppm | Fe0 (10 mg) + oscuridad | 23 | 16 min | 0.005 1/min | Gomathi-Devi et al. 2009 |
| Fe0 (10 mg) + PSA (40 ppm) + oscuridad | 47 | 0.0136 1/min | |||
| Fe0 (10 mg) + H2O2 (10 ppm) + oscuridad | 75 | 0.025 1/min | |||
| Fe0 (10 mg) + UV (125 W) | 61 | 0.0177 1/min | |||
| Fe0 (10 mg) + PSA (40 ppm) + UV (125 W) | 71 | 0.0297 1/min | |||
| Fe0 (10 mg) + H2O2 (10 ppm) + UV (125 W) | 100 | 0.1025 1/min | |||
| 20 mg/L | UV + TiO2 (0.5 g/L) | 100 | 75 min | --- | Hernández et al. 2012 |
| 80 mg/L | UV + TiO2 (0.5 g/L) | 420 min | --- | ||
| 100 mmol/L | US (20 kHz - 40 W) | 4 | 120 min | --- | Paniwnyk et al. 2010 |
| US (40 kHz - 40 W) | 1 | ||||
| US (512 kHz - 40 W) | 19 | ||||
| US (850 kHz - 40 W) | 52 | ||||
| 10 mg/L | US (40 kHz - 50 W) | 16 | 150 min | --- | Wang et al. 2005 |
| US (40 kHz - 50 W) + TiO2 (0.5 g/L) anatase | 42 | ||||
| US (40 kHz - 50 W) + TiO2 (0.5 g/L) rutilo | 97 | ||||
| 100 mg/L | MI (2450 MHz - 500 W) | 2 | 8 min | --- | Wang et al. 2011 |
| Fenton | 50 | ||||
| MI (2450 MHz - 500 W) + Fenton | 100 | ||||
| 5.4 10-5 mol/L | Fenton | --- | --- | 4.93 104 1/(M . min) | Youssef et al. 2016 |
| 20 mg/L | TiO2 SGE + oscuridad | 42 | 12 h | --- | Pang y Song 2012 |
| TiO2 SGE + UV | 50 | ||||
| 50 mg/L | TiO2 SGE + UV (15 ºC) | 29 | 0.00030 1/(% . min) | ||
| TiO2 SGE + UV (25 ºC) | 24 | 0.00098 1/(% . min) | |||
| TiO2 SGE + UV (35 ºC) | 20 | 0.00250 1/(% . min) | |||
| 0.01 mM | TiO2 SPE + oscuridad | 0 | 6 h | --- | Zhiyong et al. 2008 |
| LSS (90 mW/cm2) + PE | 8 | ||||
| TiO2 SPE + LSS (90 mW/cm2) | 100 | ||||
| TiO2 SPE + LSS (60 mW/cm2) | 90 | ||||
| LSS (90 mW/cm2) + H2O2 (1 mM) | 25 | ||||
| LSS (90 mW/cm2) + H2O2 (2 mM) | 35 | ||||
| 5 mg/L | 0.6 g/L HPA + UV (254 nm - 30 W) | 98 | 60 min | 0.0471 1/ min | Zhong 2013 |
| 10 mg/L | 80 | 0.0271 1/ min | |||
| 20 mg/L | 53 | 0.0150 1/ min | |||
| 0.3 g/L | Perreniporia tephropora (30 ºC) | 71 | 15 días | --- | Mounguengui et al. 2014 |
| 0.2 g/L | 100 | 7 días | |||
| --- | Cenizas de cáscara de arroz | 53 | 7 h | 0.02465 1/(mM . min) | Purbaningtias et al. 2015 |
| 10 mg/L | 8 mg/L Cl2O + CH (25 ºC - 0.6 MPa) | 22 | 40 min | --- | Yang et al. 2017 |
| 8 mg/L Cl2O + CH (40 ºC - 0.6 MPa) | 38 | ||||
| 8 mg/L Cl2O + CH (35 ºC - 0.4 MPa) | 71 | ||||
| 100 mg Bi2(MoO4)3 + LS + H2O2 | 100 | 70 min | 6.2 10-4 1/min | Suresh et al. 2015 | |
| 100 mg Bi2(MoO4)3 + H2O2 | 35 | 120 min | 3.3 10-5 1/min | ||
| 100 mg Bi2(MoO4)3 + LS | 5 | 60 min | 3.3 10-5 1/min | ||
| 5 mg/L | UV (12 W) + 150 mg TiO2 | 85 | 210 min | 0.0107 1/min | Koohestani y Sadrnezhaad 2016 |
| UV (12 W) + 150 mg (TiO2+ 5 % CuO) | 86 | 0.0110 1/min | |||
| UV (12 W) + 150 mg (TiO2+ 10 % CuO) | 90 | 0.0123 1/min | |||
| UV (12 W) + 150 mg (TiO2+ 15 % CuO) | 74 | 0.0075 1/min | |||
| UV (12 W) + 150 mg CuO | 40 | 0.0021 1/min | |||
| 25 ppm | TiO2 + UV | 100 | --- | --- | Kumar y Pandey 2017 |
| 100 ppm | 48 | ||||
| 1 mM | UV (365 nm - 15 W) | 2 | 170 min | --- | Kodom et al. 2012 |
| UV (365 nm - 15 W) + TiO2 / SAI | 43 | 0.00315 1/min | |||
| UV (365 nm - 15 W) + TiO2 / vidrio : SnO2: F | 58 | 0.00508 1/min | |||
| UV (365 nm - 15 W) + TiO2 / SAI + H2O2 (0.02 M) | 80 | --- | |||
| UV (365 nm - 15 W) + TiO2 / SAI + H2O2 (0.2 M) | 92 | ||||
| 20 mg/L | 0.8 g/L TiO2 (80 % anatase - 20 % rutilo) + UV (13.5 mW/cm2) | 100 | 15 min | 9.3 1/h | Marci et al. 2003 |
| 120 mg/L | 95 | 6 h | 0.6 1/h | ||
| 20 mg/L | 0.8 g/L TiO2 (anatase) + UV (13.5 mW/cm2) | --- | --- | 2.4 1/h | |
| 120 mg/L | 0.4 1/h | ||||
| 1 g/L | US (20 kHz - 70 W) + 1 g/L TiO2 (150 - 280 nm) | 62 | 1 h | --- | Ali y Abdullah 2011 |
| US (20 kHz - 70 W) + 1 g/L TiO2 (90 - 160 nm) | 82 | ||||
| 10 mg/L | UV + 1 g/L ZnO (forma cilíndrica) | 40 | 60 min | --- | Tang 2013 |
| UV + 1 g/L ZnO (forma grano de arroz) | 96 | ||||
| UV + 1 g/L ZnO (forma granular) | 99 | ||||
| 400 mg/L | US (87.5 W) + O3 (68.8 mg/L) | --- | --- | 0.24 1/min | Zhang et al. 2006 |
| US (175 W) + O3 (25.4 mg/L) | 0.15 1/min | ||||
| US (175 W) + O3 (114.2 mg/L) | 0.38 1/min | ||||
| 135.3 mg/L | US (175 W) + O3 (70.3 mg/L) | --- | --- | 0.42 1/min | |
| 3.45 mg/L | US (20 kHz - 35 W) | 17 | 45 min | 0.004 1/min | (este trabajo) |
| 3.88 mg/L | US (20 kHz - 35 W) + TM (2 mL/L) | 98 | 80 s | 1.740 1/min |
US: ultrasonido, UV: ultravioleta, PSA: persulfato de amonio, MI: microondas, SGE: soportado sobre grafito expandido, SPE: soportado sobre perileno, LS: luz solar, LSS: luz solar simulada, HPA: ácido fosfotúngstico, CH: cavitación hidrodinámica, SAI: soportado sobre acero inoxidable
En los 14 artículos revisados en los que se utilizó ultrasonido combinado con otro proceso, los porcentajes de degradación tuvieron un rango muy amplio, desde el 13 % hasta el 98 % obtenido en nuestro caso. La diferencia más notoria se presenta en los tiempos necesarios para alcanzar dichos porcentajes: en nuestro caso el mismo fue de 80 segundos, mientras que en nueve de los restantes fueron iguales o superiores a los 90 minutos (en los otros cuatro no se menciona este dato). Analizando todas las referencias en las cuales se refieren los tiempos de reacción, nuestro tiempo (80 segundos) es el menor, independientemente del proceso utilizado. Observando las constantes de decoloración puede constatarse, además, que de los 43 valores citados cinco corresponden a una cinética de segundo orden y el resto a cinéticas de primer o seudoprimer orden, donde el valor ajustado en nuestro trabajo (1.74 1/min) es el mayor.
Utilizando TM en el reactor continuo se logra una mejora en la decoloración superior al 80 % una vez que el reactor entra en estado estacionario. Mediante la simulación dinámica se encontró que los valores de las concentraciones de cada unidad coinciden con las determinadas en el ensayo cuando el reactor se encuentra en estado estacionario, pero dicho estado se alcanza en un tiempo menor al real. Esto significa que mientras el tiempo de residencia ideal es de 3.2 min, el real es mayor (cercano al doble de dicho valor).
CONCLUSIONES
Se demostró que el cloroformo puede utlizarse eficazmente como sinergizante en la decoloración sonoquímica del anaranjado de metilo, lo cual se verificó utilizando un reactor por lotes y uno continuo. En el primero se observó un aumento estadísticamente significativo en la constante de velocidad de primer orden superior a 400 veces respecto a la decoloración sin el agregado de TM. Para el caso del reactor continuo la capacidad de decoloración aumentó en más de 30 veces con el agregado de cloroformo.

