










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de ingeniería biomédica
versión On-line ISSN 2395-9126versión impresa ISSN 0188-9532
Rev. mex. ing. bioméd vol.35 no.2 México ago. 2014
Artículos de investigación original
Effects of Impaired ATP Production and Glucose Sensitivity on Human β-Cell Function: A Simulation Study
Efecto de Alteraciones en la Producción de ATP y la Sensibilidad a la Glucosa en el Funcionamiento de la Célula β Humana: un Estudio de Simulación
G.J. Félix-Martínez * J. Azpiroz-Leehan * R. Ávila-Pozos ** J.R. Godínez Fernández *
* Departamento de Ingeniería Eléctrica Universidad Autónoma Metropolitana Unidad Iztapalapa.
** Área Académica de Matemáticas y Física Universidad Autónoma del Estado de Hidalgo.
Correspondencia:
Gerardo J. Félix Martínez
Edificio AT-221 San Rafael
Atlixco 186 Col. Vicentina,
Iztapalapa C.P.09340 México, D.F.
Correo electrónico: gjfelix2005@gmail.com
Fecha de recepción: 23 de marzo de 2014
Fecha de aceptación: 15 de julio de 2014
ABSTRACT
In this paper we used a mathematical model to explore the effects of impaired ATP production and glucose sensitivity on the electrical response and insulin secretion of human β-cells. The model was extended by the addition of explicit empirical equations that describe recent experimental observations, namely, the increase of ATP as a function of glucose concentration and the oscillations in ATP at high glucose levels. Simulations were performed at selected glucose concentrations from an oral glucose tolerance test in normal subjects to evaluate the response of the human β-cell in normal and pathological scenarios. Our simulations reproduced experimental observations, such as the impaired insulin secretion as a consequence of β-cell dysfunction and restoration of electrical activity by the use of a sulfonylurea. Our results suggest that both reduced glucose sensitivity and impaired ATP production could be related to the pathogenesis of type 2 diabetes.
Keywords: action potentials, β-cell, diabetes, insulin, OGTT, calcium, ATP.
RESUMEN
En este artículo usamos un modelo matemático para explorar los efectos de alteraciones en la producción de ATP y sensibilidad a la glucosa en la respuesta eléctrica y la secreción de insulina en células β humanas. El modelo fue extendido al añadir ecuaciones empíricas explícitas que describen recientes observaciones experimentales, como el incremento en el ATP como función de la concentración de glucosa y las oscilaciones en el ATP a altos niveles de glucosa. Se realizaron simulaciones a niveles de glucosa alcanzados durante una prueba de tolerancia a la glucosa para evaluar la respuesta de la célula β humana en escenarios normales y patológicos. Nuestras simulaciones reprodujeron varias observaciones experimentales, tales como la secreción de insulina alterada como consecuencia de la disfunción de la célula β y la restauración de la actividad eléctrica al aplicar una sulfonilurea. Nuestros resultados sugieren que tanto una reducción en la sensibilidad a la glucosa como la alteración en la producción de ATP podrían estar relacionadas a la patogénesis de la diabetes tipo 2.
Palabras clave: potenciales de acción, célula β, diabetes, insulina, OGTT, calcio, ATP.
INTRODUCTION
Insulin is the only hormone capable of lowering blood glucose levels. At the systemic level, insulin inhibits the release of glucose from kidney and liver, facilitates glucose uptake in muscle and adipose tissue, and promotes the formation of glycogen in the liver[1]. Insulin is secreted by the pancreatic (β-cells, located in the islets of Langerhans, where the glucagon-, somatostatin-, and pancreatic polypeptide-producing cells (a, 5 and PP cells respectively) are also located[2]. Under normal conditions, insulin is secreted from the pancreatic β-cells by means of a well-established process known as glucose-stimulated insulin secretion (GSIS) or KATP dependent pathway: An increase in the external glucose concentration stimulates metabolism of β-cells, leading to ATP production and the closure of ATP-sensitive K+ (KATP) channels. The resulting depolarization promotes the onset of electrical activity, allowing the influx of calcium ions (Ca2+) through voltage-dependent Ca2+ channels, which eventually triggers insulin secretion (Fig. 1). It is known that GSIS is potentiated by the activation of both a neurohormonal (external) and metabolic (intrinsic) amplifying pathways[3]. The former involves the regulatory effects of hormones and neurotransmitters on insulin secretion, e.g. incretins, hormones secreted by the gastrointestinal tract that amplify the insulin response especially during oral glucose stimulation. On the other hand, the mechanisms involved in the metabolic amplifying pathway remain elusive, though it is known that a Ca2+ -independent effect of glucose on insulin secretion is involved. Furthermore, cell-cell interactions also participate in the regulation of insulin secretion, via paracrine interactions between the β, α, δ and β-cells within the islets of Langerhans[4] and the direct coupling of β-cells through gap-junctions[5].
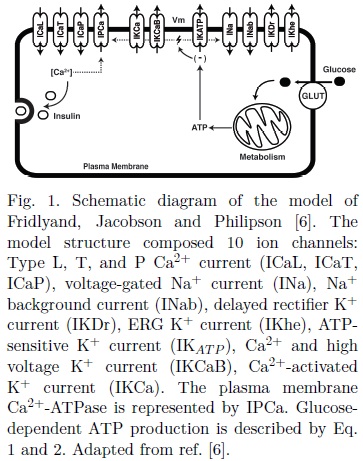
Proper functioning of β-cells is essential for glucose homeostasis, since alterations in β-cells are highly related to impaired fasting glucose (IFG) and/or impaired glucose tolerance (IGT), which eventually progress to type 2 diabetes (T2D)[7], a disease characterized by insulin resistance and β-cell dysfunction. At the cellular level, several factors could impair the adequate secretion of insulin. For example, mutations in ionic channels from human β-cell have been associated to a higher diabetes risk [9,10]. Other authors have demonstrated that a defective β-cell sensitivity and impaired metabolism could result in hyperglycemia and eventually T2D[12].
Regulation of insulin secretion has been studied extensively in rodents both experimentally and theoretically. As a complement to experimental work, mathematical models have contributed to the knowledge of β-cell physiology (reviewed in [14]). However, several important differences between human and rodent β-cells have been reported. For example, some ion channels expressed in humans are different compared to those expressed in rodents [6,10,15,16]. It is thought that these differences are responsible for the variations in the electrical behavior and secretory response. Mathematical models for human β-cells have been developed recently [6,17], aiming to analyze the mechanisms involved in the regulation of insulin secretion.
The oral glucose tolerance test (OGTT) is commonly used to assess the possible defects in β-cell function in terms of glucose sensitivity and insulin secretion [7,12,18]. In this paper we use a mathematical model and data from an OGTT in normal subjects to explore the possible causes of β-cell dysfunction in humans, one of the key aspects leading to T2D.
METHODS
Model of the human pancreatic β-cell
The model developed by Fridlyand, Jacobson and Philipson[6] was selected in this study to describe the electrical activity of human β-cells. The model was designed to evaluate the role of ionic channels in the regulation of the firing of action potentials (APs), Ca2+ dynamics and insulin secretion. Plasma membrane ion transport comprised ten ion channels (L,T and P/Q-type Ca2+ currents, voltage-gated and background Na+ currents, delayed rectifier K+ current, SK and BK Ca2+ -dependent K+ currents, ERG K+ current, ATP-sensitive K+ current) and one transporter (plasma membrane Ca2+ -ATPase). The model is shown schematically in Fig. 1.
In the original model, the relation between glucose and nucleotides concentrations was not considered. Instead, an increase in glucose was simulated by arbitrarily reducing the concentration of ADP, thus regulating the conductance of the KAT P channels.
Moreover, the affinity and inhibition constants used in the original model are based on previous models of rodent β-cells[19]. In contrast, we extended the model by deriving empirical equations from experimental observations of the production of ATP as a function of glucose concentration in human β-cells (see below) [13]. To our knowledge, the KATP channels expressed in the human β-cell have not been characterized in terms of the affinity and inhibition constants for ADP, ATP and MgADP. Values for these parameters were estimated in order to reproduce the known electrical behavior of the human βcell (i.e. the glucose threshold at which firing of APs has been detected), while maintaining the other known parameters (nucleotide total concentration and ATP basal concentration) fixed at the reported values. In addition, in our simulations the amount of MgADP was slightly reduced to 0.4ADP (0.55 ADP in the original model). Estimated and added parameters are shown in Table 1. Besides these changes, all the other parameters and formulations of the original model were adopted without modifications.

It is important to mention that both in the original and the modified model, the mechanism of insulin secretion was modeled in a minimal manner. It is known that insulin granules are distributed in distinct pools in the intracellular space, and that in response to a Ca2+ signal, granules are mobilized in order to be docked and fused with the cell membrane where the SNARE proteins (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) play a key role[20]. In mouse β-cells, it was demonstrated that the exocytotic sites are closely associated with the Ca2+ ionic channels[21]. Surprisingly, similar studies in human β-cells have not been performed. However, it is reasonable to hypothesize that a similar distribution is present in the human (cell. Recently Braun et al.[16] described the role of the Ca2+ channels in the secretory response of the human β-cell. The minimal model used in this work correctly takes into account the role of Ca2+ channels and cytosolic Ca2+ concentration in the exocytosis of insulin, though the details of the molecular machinery involved in the mobilization of insulin granules and the fusion with the cell membrane were not considered.
Glucose-induced ATP dynamics
ATP links changes in glucose metabolism to electrical activity in β-cells via the influence of ATP in the conductance of the KATP channels[8]. Early studies in human β-cells reported a glucose-dependent increase in the ATP/ADP ratio[11] which is consistent with recent experimental observations[13] that showed the relationship between glucose concentration ([G]) and the increase of sub-membrane ATP (ΔATP). This relationship was fitted to a Hill function with a half-maximal effect (KΔATP) of 5.2 mM, as reported in ref. [13]. The best fit was obtained with a Hill exponent of 5:
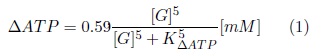
Both the experimental data and the fitted Hill function are shown in Fig. 2A. A basal ATP concentration (ATP0) of 3.5 mM and a total nucleotide concentration (NT) of 6.5 mM were assumed in accordance with the ranges reported in other studies[19].
According to experimental observations in human β-cells, high glucose ([G] > 9mM) produce oscillations in ATP, while at low glucose, oscillations were only observed occasionally[13,22]. The glucose-dependent ATP and ADP concentrations were calculated as:


For low and intermediate glucose ([G] < 9mM), we assumed the frequency of oscillations f = 0 in Eq. 2 (non-oscillatory ATP). For high glucose, Li et al. [13] reported periods of oscillation (T) of 330, 296 and 180 seconds at 9, 11 and 20 mM [G] respectively, with an approximate constant amplitude (AATP). An amplitude of 12% of the basal ATP concentration was estimated from the data reported by Ainscow et al.[22] and Li et al.[13] (AATP = 0.12ATP0).
For the high glucose scenarios, the frequency of oscillations (f in Eq. 2) was calculated as f = 2π/T. The parameter kp is a scaling factor used to simulate an impaired ATP production. The resultant glucose-dependent nucleotides concentrations (ATP0 + ΔATP) are shown in Fig. 2B. For high glucose concentrations ([G] > 9mM), oscillations generated by the second term of Eq. 2 are added to the values of ATP shown in Fig. 2B.
Oral Glucose Tolerance Test (OGTT)
The model was tested in selected glucose concentrations achieved during an oral glucose tolerance test (OGTT). Data from Ganda et al.[23] consisting in two OGTTs in 26 normal subjects were used to estimate the glucose concentrations (Fig. 2C). All simulations at selected glucose levels were performed in steady state ([G] constant).
Simulating β-cell dysfunction
Impaired glucose sensitivity was simulated by shifting the half-maximal concentration of the curve of ATP production (KΔATP, Eq. 1) to higher levels of glucose (see Fig. 5). On the other hand, a defective ATP production was simulated by decreasing the scaling factor kp in Eq. 2. Values used for KΔATP and kp are given in the figure captions. The normal physiological conditions are given by KΔATP= 5.25 mM[13] and kp= 1 (normal production of ATP).
Numerical methods
Numerical simulations were performed in Matlab, Version 2011b (MathWorks, Natick, MA). The 4th order Runge-Kutta method was used to solve the system of ordinary differential equations.
RESULTS
Onset of electrical activity
Simulations at low glucose concentrations are shown in Fig. 3. For 2 mM G, the β-cell remains electrically silent at a membrane potential of -64 mV, while [Ca2+] and insulin secretion (IS) are maintained at basal levels. As glucose is increased to 3 mM, low amplitude oscillations in Vm and [Ca2+] become apparent, with no noticeable effect on IS. Above the critical value of 3.5 mM G, low frequency action potentials emerged after a short delay, ranging from -70 to 10 mV. Changes of [Ca2+] are shown in Fig. 3C. As expected, [Ca2+] oscillates in synchrony with the changes in membrane potential, given that calcium entry depends on the activity of the voltage-gated Ca2+ channels. Low amplitude oscillations of [Ca2+] were incapable of triggering insulin secretion at 3mM G. When proper electrical activity occurred (~3.5 mM G), each action potential produced a greater increase in [Ca2+], which in turn triggered IS. Henquin et al.[24] reported a glucose threshold between ~3-4 mM for initiation of secretion in humans.

The onset of electrical activity results from the increased production of ATP at expenses of ADP as glucose levels rise, promoting the inhibition of KATP channels, membrane depolarization and activation of the voltage-dependent channels responsible for the upstroke of the APs. A threshold ATP concentration of 3.74 mM was needed to trigger electrical activity.
Electrical response of human β-cell during an OGTT
Selected input glucose levels from the OGTT curve and the corresponding ATP values are displayed in Fig. 2C. Average glucose levels during an OGTT in normal subjects range from ~3.2 mM to ~6.5 mM [23]. At low and intermediate glucose concentrations ATP remains approximately constant [13]. Eq.2 was used to calculate the ATP level, resulting in a constant glucose-dependent increase from basal ATP (Fig. 2B). Steady state simulations were performed for each input glucose both for physiological and altered conditions (Fig.4).


In normal physiological conditions (kp=1, Fig. 4A), as glucose was increased from 3.97 mM to the maximal level of 6.48 mM ([G] at t = 0 and t = 30 min during an OGTT respectively), APs showed a higher frequency, reflecte in the oscillations of IS, which is always in synchrony with the electrical activity and the changes of [Ca2+]. The amplitude of the APs remained approximately constant (not shown). Once glucose was decreased to the minimum value of ~3.29 mM ([G] after 240 min during an OGTT), insulin secretion ceased as a consequence of the repolarization of the membrane to there sting potential.
Effects of impaired production of ATP on the electrical activity of human β-cell
Impaired production of ATP was simulated by reducing ATP by a certain percentage from the calculated value (kp = 0.85, 0.74, 0.68 in Eq. 2). Reducing ATP by 15% completely inhibited electrical activity and insulin secretion at 3.97 mM G (Fig. 4A, kp= 0.85). The same results were obtained when basal ATP level was reduced by the same amount (not shown). Increasing glucose to 5.35 and 6.48 mM restored secretion, though a reduced level was observed with respect to the normal case. These results suggest that a defective production of ATP or a reduced basal ATP concentration could result in an impaired insulin secretion, one of the key characteristics of T2D. At the highest glucose level during an OGTT (6.48 mM), reducing ATP by 26% (kp= 0.74) completely inhibited electrical activity and insulin secretion (Fig. 4B). This pathological state was reverted by the action of a sulfonylurea, simulated by reducing the maximal conductance of the KATP channels (gKATP , Fig. 4B and C).
Both decreased (kp = 0.85) and full inhibited secretion (kp= 0.74) were reverted to normal levels by reducing gKATP by 20 and 33% respectively from the normal value of 45nS[6]. In Fig. 4C the restoration of the electrical activity for the case of a reduction of 26% in ATP (kp= 0.74) is shown. Stimulation of insulin secretion by tolbutamide, a commonly used sulfonylurea, has been demonstrated experimentally in human β-cells[25]. Our simulations satisfactorily reproduce these observations.
Effects of impaired glucose sensitivity on the electrical activity of human β-cells
Impaired glucose sensitivity was simulated by shifting the function of the increase of ATP (ΔATP). Fig. 5A illustrates the effect of shifting the half-maximal increase of ATP (KΔATP) from the normal value of 5.2 mM to higher glucose levels. Shifting KΔATP to 7.5mM reduced the frequency of the APs, resulting in a decreased secretion (Fig. 5B-C). On the other hand, increasing KΔATP further to 11mM inhibited the upstroke of the APs, producing only low amplitude oscillations in Vm while secretion remained at the basal level (Fig. 5B-C).
β-cell glucose sensitivity represents the dependence of insulin secretion on the glucose concentration during an OGTT[12]. In Fig. 6, simulated insulin secretion is plotted against glucose levels achieved during an OGTT. It can be seen that a shift in the half-maximal concentration of the production of ATP (KΔATP) resembles the glucose sensitivity calculated for normal, IFG and IGT subjects (compare Fig. 6 to Fig. 2A in ref. [12]).

Nucleotide oscillations and electrical activity at high glucose levels
High glucose levels are not reached during an OGTT under normal physiological conditions, however, it has been shown that in IFG and ITG subjects, glucose can achieve high concentrations (~9mM)[12]. In addition, during an intravenous glucose tolerance test (IVGTT), glucose measurements as high as ~16 mM have been reported in normal subjects[23].
According to the observations of Li et al.[13], less than 20% of the cells oscillated at 9mM G, 60% of the cells presented oscillations at 11mM G, while ~98% oscillated at 20mM G. Using these data, an oscillatory ATP concentration was calculated using Eq. 2. Simulations for the case of 9mM G are presented in Fig. 7. Oscillations of ATP between 5 and 5.8 mM produced changes in the frequency of the APs (Fig. 7D-F), which in turn caused an oscillatory insulin secretion.

Impaired production of ATP was evaluated at high glucose and oscillatory ATP (Fig. 8). Reducing ATP by 32% (kp= 0.68) produced trains of APs and a pulsatile release of insulin at glucose levels greater than 9mM. As can be seen in Fig. 8, trains of action potentials occurred when the maximal values of the oscillating ATP were achieved.

The resulting pulses of insulin had a reduced amplitude compared to the normal case (see Fig. 9). In addition, the extent of the impairment of ATP production needed to generate trains of APs was glucose-dependent. While at 9mM G a 26% decrease (kp = 0.74) on ATP generated long duration pulses of insulin secretion (Fig. 9A), reducing ATP by the same amount at 20mM G only produced a decreased level of secretion, maintaining the same oscillatory behavior than the normal case (Fig. 9B).

The pulsatile secretion caused by the trains of action potentials was reverted by the use of a sulfonylurea (Fig.10 A-B), as in the case of constant ATP. At 11 mM G, insulin pulses produced by a reduction of 32% of total ATP (kp = 0.68) returned to normal levels by decreasing gKATP by 45%, though the amplitude of the oscillations of insulin was slightly smaller compared with the normal case.
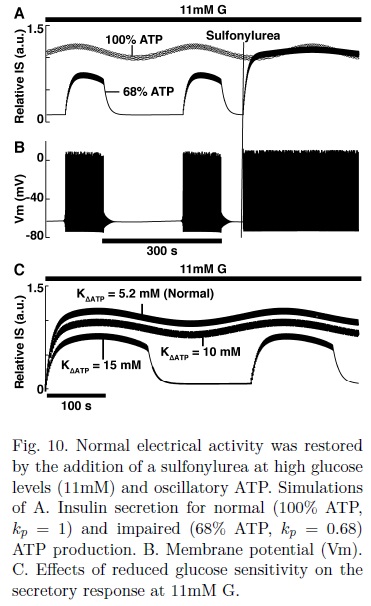
Altered glucose sensitivity at high glucose levels impaired insulin secretion but to a lesser extent in comparison to the case for low glucose and constant ATP (compare Fig. 10C and Fig. 5). As can be seen in Fig. 10C, at 11mM G, increasing KΔATP from 5.2 to 10mM only reduced insulin secretion, while at ~6.5mM G, a KΔATP of 10mM inhibited completely both electrical activity and insulin secretion (Fig. 5B). Trains of action potentials were generated for KΔATP= 15 mM, resembling the results for impaired ATP production (Fig. 9).
DISCUSSION AND CONCLUSIONS
Using a mathematical model of an isolated human β-cell we explored possible mechanisms of β-cell dysfunction under physiological and pathological conditions. In a recent publication, Fridlyand and Philipson[6] developed a model for the human β-cell including Ca2+ dynamics and a minimal model of insulin secretion. This model does not consider the relationship between glucose and metabolism explicitly. For this reason, we extended this model by adding empirical equations relating glucose and ATP production based on recent experimental data from human β-cells[13]. The proposed relations allowed us to perform simulations in various normal and pathological scenarios.
Detailed data about the relationship between glucose and electrical activity in human β-cell is lacking. Human (-cell exhibits a variable electrical activity pattern in response to a glucose stimulus. According to several studies[6,10,16], at intermediate glucose levels (~6mM), firing of individual action potentials occurs in most of the cases, while bursting is only occasionally observed. To our knowledge, the threshold glucose level for the onset of electrical activity in human β-cell is unknown. Physiological glucose levels in humans are maintained between 3 and 7 mM[10,23]; however, insulin secretion has been detected at glucose concentrations as lower as 3mM [10,26], probably as a result of a low metabolic activity maintaining a low basal rate of insulin secretion. Interestingly, experimental measurements of [Ca2+] in human β-cells showed that at 3mM G, [Ca2+] remains at the basal level[27]. Our simulations were consistent with these observations. First we evaluated the response of the model at low glucose concentrations. At 2mM, a steady basal level was observed for Vm, [Ca2+] and insulin secretion. Increasing glucose to 3mM G triggered low amplitude oscillations both in the membrane potential and [Ca2+]. The onset of electrical activity occurred at 3.5 mM G. This glucose threshold seems reasonable given that the glucose transporters found in human β-cells, GLUT1 and GLUT3[10,15,25,28], have a half maximal activity of 6 and 1mM G respectively, and glucokinase, a key enzyme in glucose metabolism have a half maximal activity at ~4mM G[29].
We can speculate that at low concentrations, glucose could be transported into the cell even at very low concentrations through GLUT 3, producing only a small amount of ATP unable to trigger electrical activity in form of action potentials. As glucose increases to higher levels, both GLUT 1 and GLUT 3 would transport glucose, accelerating metabolism further, resulting in an elevated ATP concentration, membrane depolarization and the onset of electrical activity, triggering insulin secretion.
In order to evaluate the model under realistic conditions, input glucose values were estimated from data obtained from normal subjects during an OGTT by Ganda et al.[23]. Glucose levels at t = 0, 15, 30 and 240 min, corresponding to G = 3.97, 5.35, 6.48 and 3.29 mM respectively were selected (Fig.2C). Insulin secretion increased in a glucose dependent manner as expected (Fig. 6).
Based on experimental data, Mari et al.[30] concluded that hyperglycaemia results from an intrinsic β-cell defect rather than an inadequate compensation for insulin resistance. We simulated the electrical and secretory response of a human β-cell to a glucose stimulus under pathological conditions (defective glucose sensitivity and impaired ATP production).
It has been demonstrated recently that in mouse and clonal β-cells, an impaired function of the Ca2+-selective mitochondrial uniporter (MCU) reduces the glucose-induced ATP/ADP increases [31]. This can be explained by the fact that the uptake of Ca2+ into the mitochondria activates metabolism, enhancing the production of ATP through the activation of mitochondrial enzymes[22,31]. We simulated such alterations by reducing the calculated ATP concentration (kp < 1 in Eq. 2). Our results showed that reducing ATP by 15% abolished electrical activity and insulin secretion at ~3.97mM G, while at the peak of glucose during an OGTT (6.48mM), insulin secretion was considerably impaired due to a reduced AP frequency. Application of a sulfonylurea, simulated by reducing the conductance of the KATP channels, restored the normal cell function. Based on these results, we hypothesize that a perturbation in the metabolism of β-cells (e.g. oxidative stress) could impair the adequate closure of the KATP channels, resulting in a decreased insulin secretion in human β-cells. Recently, Doliba et al.[29] showed that a defective metabolism resulting in a decreased ATP production is a key factor on the onset of TD2. Our simulations support these observations.
Oscillations of sub-membrane ATP in human β-cells were reported at high glucose levels, with a period of several minutes[13], resembling the periodicity of the well known pulsatile release of insulin in normal subjects[32]. Interestingly, in T2D, these oscillations of insulin are disrupted[32,33]. Our simulations showed that oscillations in ATP at high glucose (9, 11, 20mM) cause an oscillatory secretion with the same time period. According to the model, the cause of the oscillatory secretion is the change in frequency of the APs due to the effects of ATP in the KATP channels. When ATP production was reduced by certain amount, simulating an impaired metabolism, the average secretion level decreased (kp= 0.85), or a pulsatile secretion appeared (kp= 0.68). It should be noticed that while at 9mM G, reducing ATP by 24% (kp= 0.76) was enough to cause an impaired pulsatile secretion, at 20mM G the same effect was obtained by a greater reduction (~32%, kp =0.68).
We showed that at high glucose concentrations a decreased oscillating ATP level could produce trains of action potentials with a period of several minutes, accompanied by a decreased pulsatile insulin secretion. In contrast to rodent cells, human β-cells show a very fast bursting behavior only occasionally[10,14]. A standard model for bursting is still unknown, even for rodent β-cells. The model of Fridlyand and Philipson[6] is capable of generating a complex electrical behavior, consisting in spikes with several maximums with a period of a few seconds. This complex behavior is obtained by reducing the maximal conductance of the KATP or KCa channels. To our knowledge, there is not experimental evidence relating a reduced channel conductance with the appearance of bursting electrical activity. In contrast, the extended model here presented produced slow bursting electrical activity when an impaired oscillatory ATP was considered (Fig. 8, 9, 10) while maintaining the maximal conductances of the ionic channels unchanged. These results resemble the slow bursting pattern obtained with the model of Bertram et al. [34]. In rodent cells, both fast and slow oscillations have been observed, and several hypothesis have been proposed to explain this heterogeneous behavior (see for example ref. [14]). A feedback cycle during glycolysis resulting in an oscillatory production of ATP has been proposed as a mechanism explaining slow oscillations in rodent cells[34,35]. This proposal is consistent with experimental observations of metabolic oscillations with a similar periodicity [36,37]. On the other hand, some authors propose that the interplay between Ca2+-dependent ATP production and ATP-consuming processes have a role in the oscillations[22,38]. The periodical release of insulin could have its origin at the oscillations of ATP, via cyclic periodic changes in KATP channel activity[8]. Although oscillations in the cytosolic ATP concentration have been observed in human β-cells[13,22], more studies are needed to elucidate the mechanisms involved and the differences with other species.
Impaired glucose sensitivity is a key characteristic of T2D, and it is decreased in both IFG and IGT subjects[12]. In rat β-cells it has been associated to glucose phosphorylation rather than glucose transport [39]. Our simulations of decreased glucose sensitivity were based on this idea. We explored this pathology by shifting the curve for ΔATP to higher glucose value. At low glucose concentrations (constant ATP), shifting the curve from KΔATP = 5.2mM (normal value) to 7.5mM produced a reduced insulin secretion while KΔATP = 10 completely inhibited electrical activity and insulin secretion. On the other hand, at high glucose levels (oscillatory ATP), the same scenarios caused a noticeable but small impairment on insulin secretion. In fact, only shifting KΔATP to 15mM inhibited normal functioning.
It is worth noting that the extended model only includes the mechanisms involved in the KATP-dependent pathway in an isolated human β-cell, thus omitting other regulatory processes. An important extension to the model would be to incorporate the effect of incretins such as glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptid (GIP), which are known to potentiate the secretory response during oral glucose estimulation (as in an OGTT)[40]. This would allow us to address the known differences in the secretory response during oral and intravenous stimulation, namely, that oral glucose produces a greater secretory response in comparison to intravenous glucose[41]. On the other hand, both the original and the extended model use a minimal model to simulate the exocytosis of insulin granules. A more detailed model could be incorporated in future work to describe in more detail the mechanisms of cell secretion, including the mobilization to the cell membrane and exocytosis of insulin granules in β-cells.
In the extended model presented here, simulations assuming a steady state were performed. In other words, we have simulated the response of the isolated human β-cell due to a constant glucose stimulus estimated from the plasma glucose concentration curve following an OGTT.
Future modeling work should be done using a multiscale approach in order to incorporate both cell-cell interactions and crucial systemic processes (e.g. glucose uptake by muscle an adipose tissue, the effects of insulin on liver and kidney and the neurohormonal amplifying pathway), which will allow us not only to evaluate the function of the β-cell dinamically, but also to simulate the response of the glucose-insulin system in the healthy, prediabetic and diabetic states.
We can conclude that the proposed relationship between glucose and ATP production, in conjunction with the model of human β-cells of Fridlyand, Jacobson and Philipson[6] was able to reproduce several experimental observations, both in normal and impaired conditions. The results of the simulations showed that at the cellular level, both reduced glucose sensitivity and impaired ATP production could be related to the pathogenesis of T2D.
REFERENCES
1. M.Z. Shrayyef, J.E. Gerich, Normal glucose homeostasis, In: Principles of Diabetes Mellitus, Springer US, 2009: pp. 19-35. [ Links ]
2. M. Brissova, Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy, J Histochem Cytochem. 53 (2005) 1087-1097. [ Links ]
3. J.C. Henquin, The dual control of insulin secretion by glucose involves triggering and amplifying pathways in β-cells, Diabetes Res Clin Pr. 93 (2011) S27-S31. [ Links ]
4. D.S. Koh, J.-H. Cho, L. Chen, Paracrine interactions within islets of Langerhans, J Mol Neurosci. 48 (2012) 429-440. [ Links ]
5. J.V. Rocheleau, M.S. Remedi, B. Granada, et al., Critical role of gap junction coupled KATP channel activity for regulated insulin secretion, PLoS Biol. 4 (2006) e26. [ Links ]
6. L.E. Fridlyand, D.A. Jacobson, L.H. Philipson, Ion channels and regulation of insulin secretion in human β-cells: a computational systems analysis, Islets. 5 (2013) 1-15. [ Links ]
7. A. Basu, M.G. Pedersen, C. Cobelli, Prediabetes: Evaluation of β-Cell Function, Diabetes. 61 (2012) 270-271. [ Links ]
8. S. Dryselius, P.E. Lund, E. Gylfe, B. Hellman, Variations in ATP-Sensitive K+ channel activity provide evidence for inherent metabolic oscillations in pancreatic β-cells, Biochem Biophys Res Commun. 205 (1994) 880-885. [ Links ]
9. A.H. Rosengren, M. Braun, T. Mahdi, et al., Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes, Diabetes. 61 (2012) 1726-1733. [ Links ]
10. P. Rorsman, M. Braun, Regulation of insulin secretion in human pancreatic islets, Annu Rev Physiol 75 (2013) 155-179. [ Links ]
11. P. Detimary, S. Dejonghe, Z. Ling, D. Pipeleers, F. Schuit, J. Henquin, The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in β cells but not in cells and are also observed in human islets, J Biol Chem. 273 (1998) 33905-33908. [ Links ]
12. M. Kanat, A. Mari, L. Norton, et al., Distinct β-cell defects in impaired fasting glucose and impaired glucose tolerance, Diabetes. 61 (2012) 447-453. [ Links ]
13. J. Li, H.Y. Shuai, E. Gylfe, A. Tengholm, Oscillations of sub-membrane ATP in glucose-stimulated beta cells depend on negative feedback from Ca2+, Diabetologia. 56 (2013) 1577-1586. [ Links ]
14. L.E. Fridlyand, N. Tamarina, L.H. Philipson, Bursting and calcium oscillations in pancreatic beta-cells: specific pacemakers for specific mechanisms, Am J Physiol Endocrinol Metab. 299 (2010) E517-32. [ Links ]
15. M. Hiriart, L. Aguilar-Bryan, Channel regulation of glucose sensing in the pancreatic β-cell, Am J Physiol Endocrinol Metab. 295 (2008) E1298-306. [ Links ]
16. M. Braun, R. Ramracheya, M. Bengtsson, et al., Voltage-gated ion channels in human pancreatic β-cells: electrophysiological characterization and role in insulin secretion, Diabetes. 57 (2008) 1618-1628. [ Links ]
17. M. Riz, M. Braun, M.G. Pedersen, Mathematical modeling of heterogeneous electrophysiological responses in human β cells, PLoS Comp Biol. 10 (2014) e1003389. [ Links ]
18. E. Ferrannini, A. Gastaldelli, Y. Miyazaki, M. Matsuda, A. Mari, R.A. DeFronzo, β-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis, J Clin Endocrinol Metab. 90 (2005) 493-500. [ Links ]
19. L.E. Fridlyand, L. Ma, L.H. Philipson, Adenine nucleotide regulation in pancreatic β-cells: modeling of ATP/ADP-Ca2+ interactions, Am J Physiol Endocrinol Metab. 289 (2005) E839-48. [ Links ]
20. S. Nagamatsu, M. Ohara-Imaizumi, Mechanism of insulin exocytosis analyzed by imaging techniques, In: Pancreatic Beta Cell in Health and Disease, Springer Japan, 2008: pp. 177-193. [ Links ]
21. K. Bokvist, K., Eliasson, L., Ammálá, C., Renstrom, E., Rorsman, P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. EMBO J, 14(1) (1995) 50-57. [ Links ]
22. E.K. Ainscow, G.A. Rutter, Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet β-cells: evidence for a Ca2+-dependent mechanism, Diabetes. 51 Suppl 1 (2002) S162-70. [ Links ]
23. O.P. Ganda, J.L. Day, J.S. Soeldner, J.J. Connon, R.E. Gleason, Reproducibility and comparative analysis of repeated intravenous and oral glucose tolerance tests, Diabetes. 27 (1978) 715-725. [ Links ]
24. J.C. Henquin, D. Dufrane, M. Nenquin, Nutrient control of insulin secretion in isolated normal human islets, Diabetes. 55 (2006) 3470-3477. [ Links ]
25. S.G. Straub, R.F. James, M.J. Dunne, G.W.G. Sharp, Glucose activates both KATPchannel-dependent and KATP channel-independent signaling pathways in human islets, Diabetes. 47 (1998) 758-763. [ Links ]
26. M. Braun, R. Ramracheya, P.R. Johnson, P. Rorsman, Exocytotic properties of human pancreatic β-cells, Ann NY Acad Sci 1152 (2009) 187-193. [ Links ]
27. I. Quesada, M.G. Todorova, P. Alonso-Magdalena, et al., Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified α- and β-cells within intact human islets of Langerhans, Diabetes. 55 (2006) 2463-2469. [ Links ]
28. L.J. McCulloch, M. van de Bunt, M. Braun, GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus, Mol Genet Metab. 104 (2011) 648-653. [ Links ]
29. N.M. Doliba, W. Qin, H. Najafi, et al., Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics, Am J Physiol Endocrinol Metab. 302 (2012) E87-E102. [ Links ]
30. A. Mari, A. Tura, A. Natali, et al., Impaired beta cell glucose sensitivity rather than inadequate compensation for insulin resistance is the dominant defect in glucose intolerance, Diabetologia. 53 (2010) 749-756. [ Links ]
31. A.I. Tarasov, F. Semplici, M.A. Ravier, et al., The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells, PloS One. 7 (2012) e39722. [ Links ]
32. A. Tengholm, E. Gylfe, Oscillatory control of insulin secretion, Mol Cell Endocrinol 297 (2009) 58-72. [ Links ]
33. P. Bergsten, Pathophysiology of impaired pulsatile insulin release, Diabetes Metab Res Rev. 16 (2000) 179-191. [ Links ]
34. R. Bertram, L.S. Satin, M.G. Pedersen, D.S. Luciani, A. Sherman, Interaction of glycolysis and mitochondrial respiration in metabolic oscillations of pancreatic islets, Biophy J. 92 (2007) 1544-1555. [ Links ]
35. M.J. Merrins, R. Bertram, A. Sherman, L.S. Satin, Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase modulates oscillations of pancreatic islet metabolism, PloS One. 7 (2012) e34036. [ Links ]
36. R.T. Kennedy, L.M. Kauri, G.M. Dahlgren, S.K. Jung, Metabolic oscillations in β-cells, Diabetes. 51 Suppl 1 (2002) S152-61. [ Links ]
37. D.S. Luciani, Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets, J Physiol. 572 (2006) 379-392. [ Links ]
38. P. Detimary, P. Gilon, J. Henquin, Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets, Biochem J. 333 (Pt 2) (1998) 269-274. [ Links ]
39. H. Heimberg, A. De Vos, A. Vandercammen, E. Van Schaftingen, D. Pipeleers, F. Schuit, Heterogeneity in glucose sensitivity among pancreatic β-cells is correlated to differences in glucose phosphorylation rather than glucose transport, Embo J. 12 (1993) 2873-2879. [ Links ]
40. S. Klinger, B. Thorens, Molecular Biology of Gluco-Incretin Function, In: Pancreatic Beta Cell in Health and Disease, Springer Japan, 2008: pp. 315-334. [ Links ]
41. E.R. Pearson, I. Flechtner, P.R. Njølstad, et al., Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations, N Engl J Med. 355 (2006) 467-477. [ Links ]

