










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.3 Ciudad de México jul./sep. 2002
Investigación
Evidencias espectroscópicas de RMN de 1H y 13C en la formación inusual de un "carbonilo inorgánico", derivado de la lactona costunólida
Eduardo Díaz,*a Héctor Barrios,a Aydeé Fuentes,b David Coronaa y Ángel Guzmána
a Instituto de Química, Universidad Nacional Autónoma de México. Ciudad Universitaria Circuito Exterior. Coyoacán 04510, México D.F. Teléfono: 5622-4421.
b Facultad de Química. Universidad Autónoma del Estado de México Paseo Colón y Tollocán; Toluca E. de México CP 50000.
Recibido el 6 de mayo del 2002.
Aceptado el 26 de agosto del 2002.
Resumen
El uso de la RMN de 1H y 13C permitió determinar la estructura inusual de un "carbonilo inorgánico" derivado de la lactona dehidrocostus.
Palabras clave: RMN de 1H y 13C, derivados de productos naturales.
Abstract
1H and 13C NMR of an unusual "inorganic carbonyl" derived from costunolide lactone are described.
Keywords: 1H and 13C NMR. Natural products derivatives.
Dedicado al Dr Barbarín Arreguín por toda una vida en la actividad científica
Introducción
Los compuestos orgánicos aislados de productos naturales ofrecen una abundante gama de estructuras que contienen grupos funcionales muy variados. Estos constituyen objetivos interesantes para poder efectuar un gran número de reacciones químicas que permitan introducir y transformar diferentes funcionalidades en la molécula.
De estos productos naturales, las lactonas sesquiterpénicas han sido objeto de estudios muy variados sobre la utilización y funcionalización del metileno exocíclico de la lactona, o también sobre algún otro grupo funcional presente en la estructura. Siguiendo este criterio, se han logrado obtener derivados dibromociclopropánicos [1], ciclopropánicos [2], o bien derivados de uracilos usando reacciones de Michael, las cuales demostraron efectuarse con características estereoselectivas [3-5].
Los derivados dibromociclopropánicos con los que se han funcionalizado las estructuras de las lactonas sesquiterpénicas, han servido como intermediarios para acceder a sistemas alénicos a través de la bien conocida reacción de Hiyama [6].
Entre las investigaciones más recientes efectuadas por nuestro grupo, están las reacciones con diclorocarbenos [7], usando técnicas de transferencia de fase con NaOH, CHCl3 y un catalizador conocido como TEBAC.
Ahora, en este trabajo describimos las estructuras de dos subproductos aislados de la reacción de la lactona costunólida con CHCl3 / NaOH / TEBAC y con CHCl3 / NaOH / Arquad / Acetona respectivamente, los cuales presentan comportamientos inusuales y cuyas características estructurales fueron elucidadas a través de RMN de 1H y 13C, apoyadas por la información obtenida de espectrometría de masas.
Discusión y resultados
La reacción de dihalocarbenación con reactivos de Seyferth [8] se ha usado ampliamente para obtener dihalociclopropanos. Esta reacción permite a partir de sustratos estables acceder a productos con estructuras bien definidas.
En el caso de derivados dibromociclopropánicos o diclorociclopropánicos, los reactivos de Seyferth se han usado exitosamente. Sin embargo, debido a que en la transformación es necesario usar temperaturas de reflujo del disolvente usado en la reacción, este proceso resulta inadecuado en los casos en los que la materia prima se descompone o polimeriza. Tal es el caso de la lactona costunólida 1; por lo tanto, es necesario buscar una reacción alternativa para evitar tales inconvenientes.
Las reacciones de transferencia de fase han constituído un método ideal para las reacciones de dihalocarbenación aplicados a sistemas lábiles.

En el caso de la lactona costunólida 1, cuando es tratada con CHBr3 / NaOH / Arquad, se obtuvieron mono, bis y tris dibromociclopropanos [9]. Sin embargo, cuando la reacción se efectuó usando la misma lactona con CHCl3 / NaOH / TEBAC o bien con CHCl3 / NaOH / Arquad / acetona no se obtuvo ningún derivado diclorociclopropánico, obteniéndose en su lugar dos productos (2 y 3) cuyas estructuras constituyen el objeto de este trabajo.
Cuando el compuesto 1 se puso a reaccionar con CHCl3 / NaOH / TEBAC, se aisló un sólido cristalino de punto de fusión 160-163 °C cuyo espectro de resonancia magnética nuclear protónica mostró los hidrógenos del metileno exocíclico (H-13 y H-13') en δ = 5.21 y 4.76 (DMSO-d6, 500 MHz; Figs. 1 y 2).


Dicha observación solo puede ser explicada por una pérdida en la conjugación del grupo α,β insaturado de la γ lactona debido a la desaparición del carbonilo C-12. La ausencia de este grupo fue fácilmente detectada en el espectro de IR (véase parte experimental) y en la lectura en el espectro de masas (FAB) de un ion molecular m/z = 293 que sugiere la inclusión de dos átomos de sodio y un oxígeno en el esqueleto de la γ lactona.
El espectro de RMN de 13C mostró una señal en δ = 174.0. La forma para complementar dicha observación con lo detectado en RMN de 1H e IR, solo puede ser posible asumiendo la formación de un "carbonilo inorgánico" [10-11] como se representa en la estructura 2. Estos carbonilos están estrechamente relacionados con los iones oxocarbonio estables y que son intermediarios de gran interés en virtud de que se ha observado que ácidos carbónicos protonados han mostrado ser agentes carboxilantes efectivos y juegan un papel importante en ciertos procesos biológicos de carboxilación.
Asimismo, las observaciones espectroscópicas de este tipo de intermediarios han sido exitosamente efectuadas usando muy bajas temperaturas (-40 a -80 °C) y en presencia de mezclas conocidas como superácidos (FSO3H-SbF5-SO2).
Olah [12] ha informado la obtención de 2-hidroxi-1,3-dioxolium hexafluoro antimoniato a partir de una solución de carbonato de vinileno disuelto en HF-SbF5-SO2. La sal es un material blanco cristalino, higroscópico y que en presencia de aire seco se oscurece a temperatura ambiente.


De lo anteriormente expuesto se observa que la posibilidad de obtener los llamados "carbonilos inorgánicos" bajo condiciones menos severas no es algo usual.
Por otro lado, cuando la reacción se efectuó sobre la lactona costunólida usando CHCl3 / NaOH / Arquad / acetona, se obtuvo un nuevo producto (3) derivado de la adición de Michael del enolato de la acetona sobre el metileno exocíclico C-11- C-13.
La estructura del compuesto 3 se elucidó por la observación en la RMN de 1H de la desaparición de las señales debidas al metileno exocíclico presente en la lactona costunólida (δ = 6.04 y 5.64) observándose en su lugar las señales de un nuevo metileno saturado en δ =1.76 y 1.96 y la aparición de un singulete (3H) en δ = 2.17 correspondiente a una metil cetona. En el espectro de IR dos bandas de grupo carbonilo fueron detectadas; 1763 cm-1 (γ-lactona) y 1713 cm-1 cetona). En el espectro de masas la observación de un ion molecular (M+) m/z 288 sugiere la adición de 58 unidades de masa (C3H6O) al metileno exocíclico de la lactona costunólida tal como se representa en 3 (Fig. 3). Los datos completos de RMN de los desplazamientos químicos de 1H y 13C del compuesto 3, así como sus correlaciones heteronucleares (HMBC) están representados en la Tabla 1.
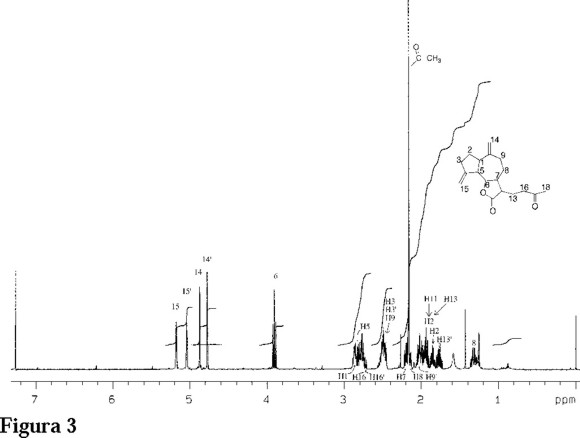
La evidencia del centro estereogénico generado por la adición de la acetona sobre el C-13 fue deducida de la constante de acoplamiento 3JH-11-H-7 = 9.5 Hz. La magnitud de este acoplamiento solo puede ser observada si H-7 y H-11 están en una geometría anti [13]. Esta consideración está de acuerdo con las observaciones anteriores hechas para la misma lactona [5].
Para explicar porqué la adición de Michael del enolato de la acetona sobre el metileno exocíclico se efectúa por la cara α se llevó a cabo una comparación energética por medio de modelaje molecular sobre las moléculas representadas en el esquema 3.
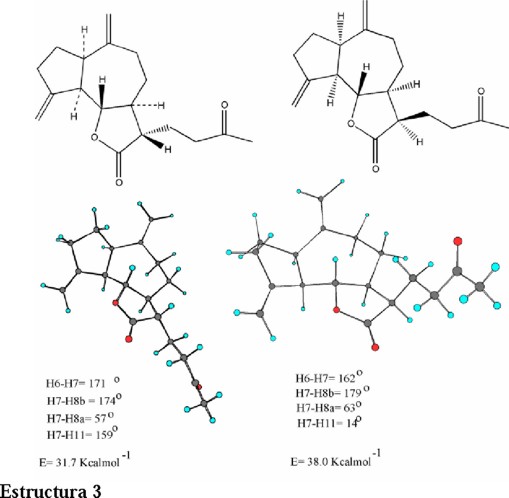
La diferencia de 6.3 Kcal mol-1 entre ambas estructuras fácilmente permite deducir que cuando el grupo adicionado al metileno exocíclico es β, una interacción synperiplanar entre C-13 / C-8 se efectúa y por lo tanto esta configuración presentará una mayor energía estérica que aquella estructura que presenta la cadena unida al C-13 en posición α. Esta predicción está de acuerdo con lo observado experimentalmente a través de la constante de acoplamiento vecinal H7-H11.
Parte experimental
Los espectros de masas fueron determinados en instrumentos JEOL JMS-AX505 HA y JEOL-JMS-10217 usando impacto electrónico, ionización química o FAB. Los espectros de infrarrojo fueron detectados en instrumentos Nicolet FX-SX y Nicolet 55-X.
Los espectros de alta resolución se determinaron en un instrumento Varian Unity 500 operando a 500.3 MHz para 1H y 125.0 MHz para 13C. Los experimentos fueron efectuados usando una sonda de detección inversa de 5 mm. Los experimentos de HMQC y HMBC se corrieron usando el programa estándar habitual de Varian Unity. Los espectros de los compuestos 1 y 2 fueron obtenidos en solución de DMSO-d6 usando TMS como referencia interna (0.00 ppm). El compuesto 3 se analizó en solución de cloroformo deuterado y TMS como referencia.
Procedimientos generales
A una solución de 200 mg de lactona dehidrocostus en 1.5 mL de cloroformo, se agregaron 6.0 mL de solución de hidróxido de sodio (10 %) y 20 mg de TEBAC. La reacción se agitó durante 24 h monitoreándola por cromatografía en capa fina. Después del trabajo usual el residuo se purificó por cromatografía en columna (flash, silica gel, hexano / acetato de etilo 9:1) para dar el derivado 2.
El compuesto 2 presentó un pf = 160-163 °C ; [RMN 1H, δ ppm; 5.21, 5.08, 4.81, 4.76, 4.71, 4.59, 3.21, 2.74, 2.49, 2.52, 2.36, 2.33, 2.26, 2.00, 1.86, 1.70, 1.67, 1.47].
NMR 13C δ ppm: 174.0, 154.2, 153.2, 152.2, 112.6, 109.1, 107.5, 74.1, 55.7, 50.8, 46.4, 36.3, 34.5, 31.3, 29.1.
MS, FAB: M+ m/z 293 (100), C15H18O3Na2; m/z 271 (55); 254 (20), 176 (80), 154 (40), 101 (60), 79 (80).
IR (KBr) νmax cm-1; 3320, 2933, 1677, 1547, 1427, 1383.
A una solución de 1.0 g de lactona dehidrocostus en 15 mL de cloroformo, 5 mL de acetona y 100 mg de Arquad S-50 se le fue agregando gota a gota 7.5 mL de una solución de hidróxido de sodio (50 %). La solución tomó una coloración amarilla. Se dejó agitar a temperatura ambiente por 24 h. Después del trabajo usual, el residuo se purificó por cromatografía en placa preparativa dando unos cristales blancos de pf 168-169 °C que fueron identificados como el derivado 3.
[El compuesto 3 presentó un espectro de RMN de 1H ppm: 5.18, 5.05, 4.87, 4.78, 3.90, 2.86, 2.83, 2.74, 2.49, 2.48, 1.96, 2.20, 2.17, 2.15, 2.04, 2.02, 1.96, 1.94, 1.84, 1.76, 1.32. RMN 13C δ ppm: 208.1, 177.9, 151.6, 149.8, 111.9, 109.2, 85.1, 52.0, 47.8, 47.1, 45.5, 40.0, 37.4, 32.4, 30.1, 30.0, 22.2].
MS, IE: M+ m/z 288, C18H24O3, M+ -17, m/z 270; m/5 158; m/z 43 (100).
IR, νmax cm-1: 3020, 2927, 2857, 1763, 1713, 1448, 1215, 1174, 760.
Agradecimientos
Al Ing. L. Velasco y al M.C. J. Pérez por los E.M.; Quím. R. Patiño por los espectros de IR y a la M.C. M.I. Chávez por los espectros de RMN de 1H y 13C.
Bibliografía
1. Toscano, R.A.; Nava, J.L.; Guerrero, C.; Guzmán, A.; Díaz, E. J. Chem. Crystallography 1997, 27, 457-463. [ Links ]
2. Díaz, E.; Dominguez, G.G.; Mannino, A.; Negrón, G; Jankowski, K. Magn. Reson. Chem. 1985, 23, 494-502. [ Links ]
3. Díaz, E.; Barrios, H.; Nava, J.L.; Enriquez, R.G.; Guzmán, A.; León, L.; Fuentes, J.F.; Fuentes, A.; Quintero, A.; SolanoJ.D. J. Heterocyclic. Chem. 1997, 34, 1037-1041. [ Links ]
4. Díaz, E.; Nava, J.L.; Barrios, H.; Quiroz, B.; Guzmán, A.; León, L.; Fuentes, A. Spectrochimica Acta. 1998, Part A, 54, 567-574. [ Links ]
5. Díaz, E.; Barrios, H.; Nava, J.L.; Chávez, M.I.; Guzmán A.; Fuentes, J.F.; Fuentes, A.; Spectroscopy Lett. 1998, 31, 51-61. [ Links ]
6. a) Díaz, E.; Ortega, A.; Guerrero, C.; Guzmán, A.; Lizama, B.E.; Jankowski, K. Natural Products Lett. 1998, 12, 241-254. [ Links ] b) Hiyama, T.; Okude, Y.; Kumura, I.K.; Nozaki, N. Bull. Chem. Soc. Japan 1982, 55, 561-568. [ Links ]
7. a) Díaz, E.; Toscano, R.A.; Alvarez, A.; Shoolery, J.N.; Jankowski, K. Can. J. Chem. 1990, 68, 701-704. [ Links ] b) Díaz, E.; Fuentes, A.; Villafranca, E.V.; Jankowski, K. Acta Crystallographica 1994, C-50, 2030-2032. [ Links ]
8. a) Seyferth, D. Accounts Chem. Res. 1972, 5, 561. [ Links ] b) Seyferth, D; Mui, J.Y.P; Burlich, J.M. J. Amer. Chem. Soc. 1967, 89, 4953-4959. [ Links ] c) Seyferth, D.; Burlich, J.M. J. Amer. Chem. Soc. 1964, 86, 2730-2731. [ Links ]
9. Díaz, E., resultados aún no publicados.
10. Stothers, J.B. en: C-13 NMR spectroscopy, Academic Press. N.Y. 303-305 1972. [ Links ]
11. Le Quesne, P.W.; Levery, S.B.; Menachery, M.D.; Brennan, F.F.; Raffault, R.F. J.Chem. Soc. Perkin Trans. I 1978, 1572-1580. [ Links ]
12. a) Olah, G.A.; White, A.M. J. Amer. Chem. Soc. 1967, 89, 7072-7075. [ Links ] b) Olah, G.A.; White, A.M. J. Amer. Chem. Soc. 1968, 90, 1884-1889. [ Links ]
13. a) Karplus, M. J. Amer. Chem. Soc. 1963, 85, 2870-2871. [ Links ] b) Bhacca, N.; Williams, D.H. in: Applications of NMR Spectroscopy in Organic Chemistry, Holden Day, San Francisco, 1964. [ Links ]

