










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.47 no.2 Ciudad de México abr./jun. 2003
Investigación
2D 1H and 13C NMR from the adducts of the dichloro carbene addition to β-ionone. The role of the catalyst on the phase transfer reaction
Eduardo Díaz,*a José Luis Nava,a Héctor Barrios,a David Corona,a Ángel Guzmán,a Ma. de Lourdes Muciño,b and Aydeé Fuentesb
a Instituto de Química, Universidad Nacional Autónoma de México. Circuito Exterior, Ciudad Universitaria, Coyoacán 04510 México D.F.
b Facultad de Química, Universidad Autónoma del Estado de México, Toluca, Estado de México.
Recibido el 9 de diciembre del 2002.
Aceptado el 26 de febrero del 2003.
En homenaje al Dr. Alfonso Romo de Vivar.
Abstract
Several haloderivatives were synthesized from β-ionone using CHCl3, NaOH and selected tetralkyl ammonium halide. 2D NMR and X-ray single crystal analysis of the products are reported.
Keywords: Addition of dichlorocarbenes to β-Ionone, phase transfer addition, formation of 1,1 dichlorocyclopropanes. 1H and 13C NMR.
Resumen
Algunos haloderivados fueron sintetizados a partir de β-ionona usando cloroformo, hidróxido de sodio y halogenuros de tetralquilamonio selectos. Se informan los análisis por 2D RMN y rayos X de los productos.
Palabras clave: Adición de diclorocarbenos a β-ionona, adición de transferencia de fase, formación de 1,1 diclorociclopropanos. RMN de 1H y 13C.
Introduction
A large number of C13-compounds formed by oxidative cleavage of carotenoids have been isolated from tobacco [1] and marine sponges [2] or their derivatives, originated from α and β ionones (1' and 1). In the same way, some other secondary metabolites as 4-oxomegastigmenos, important compounds for their excellent flavor characteristics are also formed in the tobacco plant from α and β ionones. Other derivatives from ionones, isolated from rabbit urine and from the secretion of the anal gland of the red fox have been obtained when α-ionone was photo-oxygenated [3, 4].
The synthesis of several damascones [5] and abscisic acids [6, 7] using readily available and inexpensive ionones as raw material encourages us to perform a reaction leading to some new functionalized and useful unsaturated compounds.
Results and discussion
The present paper deals with the synthesis and structure elucidation of new representatives of this group of compounds. We describe here the products obtained when β-ionone was treated with CHCl3, NaOH, using as catalyst different tetralkyl quaternary ammonium salts looking to improve yields and versatility of the reaction under phase transfer conditions.
Likewise, the reaction of carbenes with terpenes provides a simple means of examining the stereochemistry and regioselectivity of addition of such species to a variety of double bonds.
It has been reported that in phase transfer dihalocarbene addition, stereochemistry and regioselectivity can be controlled by varying the catalyst [8-11]. Phase transfer reactions are known to be somewhat dependent on the exact reaction conditions and the formation of products apparently derived from trihalogenomethyl anion or dihalogen carbene can be controlled by varying the catalyst [9].
Some years ago [12], our group reported β-Ionone when treated under CHCl3, NaOH and triethylbenzyl ammonium chloride (TEBAC) yield two furenones 2 and 3 which were isolated in low yield. Their structures were well supported by 2D NMR as well as x-ray crystallographic analysis [13].
On the mechanism of formation of these compounds it was assumed an initial chemoselective adduct formation on the β-ionone to generate the epoxide, which is rapidly transformed to an intermediate dichloro ether which is hydrolized to a γ-lactone where the presence of atmospheric oxygen during the reaction induced free radical dimerization [12].
The low yield observed in this reaction as well as the new findings about the phase transfer reactions encourage us to undertake the study of the dichlorocarbene addition to β-ionone using different quaternary ammonium catalyst.
Under the new catalyst selection, the usual γ-lactone derivatives (2 and 3) were isolated together with several new compounds which display structure versatility and in addition allowed us to improve yields, as well as products ratio of the obtained adducts.
Structures of compounds 2 and 3 were well discussed in our previous report and they will not be focused here.
Scheme 1 shows the structures of the new isolated derivatives.
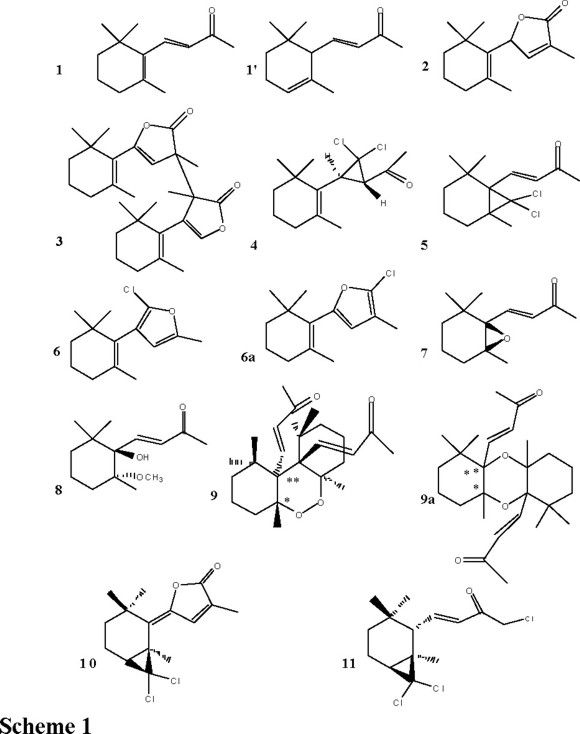
The IR spectrum of 4 showed an absorption at 1717 cm−1 which clearly indicates an unsaturated ketone group. The proton NMR spectrum showed a one proton doublet and one proton multiplet at δ = 3.04 and 2.83 respectively. The latter was converted to a doublet of triplet when the methyl protons (C7) were decoupled. A methyl ketone group appeared at δ = 2.45 as a singlet. These findings suggest that in the structure of compound 4 was lost the α,β-unsaturated double bond observed in the starting material 1. The mass spectrum of compound 4 shows the molecular ions at m/z 274 (M+), m/z 276 (M++ 2) and m/z 278 (M+ + 4) corresponding to the molecular formula of C14H20OCl2. It suggests an increase in molecular weight of the starting ionone 1 by 82 atomic mass units (CCl2).
The NOESY experiment enabled us to establish the key proton vicinities. There exist a strong correlations between methine doublet at δ = 3.04 and methyl signals δ = 2.45 (C-13), 1.00 (C-9) and a weaker one between methyne multiplet at =2.83 and the methyl singlet at δ = 1.75.
The 13C 1D NMR and DEPT edited spectra [14] allowed the assignment of the carbon atoms (protonated carbons), and the long range heterocorrelated spectra [15] enabled us to establish connectivities between protons and carbons (see experimental).
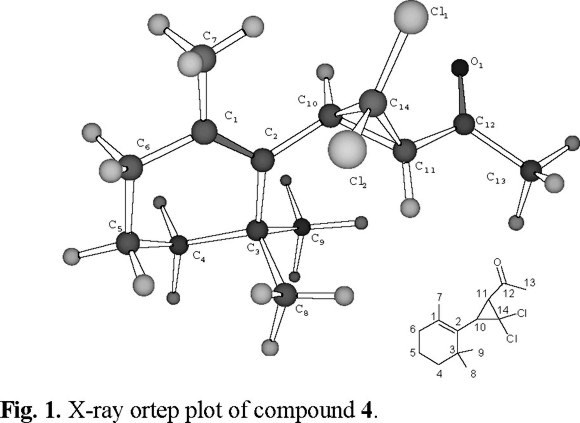
Because the unusual trans cyclopropane vicinal coupling between HC10-HC11 (J = 9.0 Hz) and in order to confirm this adduct structure we carried out an x-ray crystallographic study of compound 4. Fig. 1 display the Ortep plot of compound 4.
On the other hand, compound 5 shows a molecular weight as was identified from MS as 274, 276, 278 (C14H20O Cl2). The proton NMR presents an AX pattern (δA = 6.63 (H-10) and δX = 6.17 (H-11, J=16.0 Hz), four singlets at δ = 2.30, 1.25, 1.20 and 1.00 for the methyls at C-13, C-7, C-8 and C-9 and the signals for six protons on C-4, C-5 and C-6 methylenes. The structure of compound 5 supported by the assignments of 1H and 13C (CDCl3, 500 MHz , 125 MHz) spectra and confirmed by HMQC, HMBC along with HOMOCOSY and the mass spectrum fragmentation pattern matching the expected molecular weight for such a structure.
The structure of the monochlorine furane derivative 6 was supported by its spectroscopic features. The molecular ion M+ was observed in mass spectrometry (EI) at m/z 238 (M+), m/z 240 (M+ + 2) in agreement with the molecular weight for a furane derivative. Tentatively, two isomeric structures (6 and 6a) emerge to be considered under mechanistic approach (Scheme 2).

The 1H and 13C NMR (CDCl3, 300 MHz, 75.0 MHz), together with HETCOR [16, 17] and COLOC [18] spectra were recorded in order to probe the structure 6. At first, since 13CNMR chemical shifts observed (and calculated) [19] for 6 (or 6a) did not enable unambiguously to differentiate between 6 or 6a, we performed a COLOC experiment in order to overcome such drawback. In this experiment we were able to observe the long range proton-carbon correlation between the furane methyl protons (δ = 2.20, C-13) with the methine C-11 (δ = 111.0) and with the nonprotonated carbon at δ = 149.9 (C-12). It suggests unambiguously structure 6 for this furane derivative. In the structure 6a the 3σ bond correlation between the methyl group with the carbon at δ = 149.9 is not permissible (Fig. 2).
The remaining signals that enabled us the elucidation of structure 6 are described in experimental.
On the other hand, the formation of the dimeric compound 9 (or 9a, 9a' or 9') can be explained by two different mechanism approach. At first, if we assume that compound 9a (or 9a') could be formed by a concerted opening and dimerization of the epoxide 7 (Scheme 3) obtained in a small amount under phase transfer conditions. This assumption was considered because the previously mentioned opening was for us described and probed, since we were able to isolate the alcohol-methoxyether derivative 8, whose structure and stereochemistry were confirmed by an x-ray crystallographic study [12].

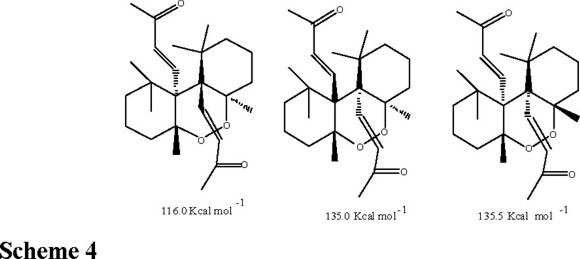
The 13C NMR chemical shifts were a key element in order to rule out the structures of the dioxine derivatives 9a, 9a' or the oxide 9'. The calculated chemical shifts [19] for the non protonated carbons attached to both oxygen atoms (** and * in schemes 1 and 3) should to display chemical shifts at δ = 90.0 and 71.0, respectively. However, compound 9 showed in its 13C NMR chemical shifts at δ =71.0 and 65.8 ppm respectively, which definitely support the structure 9 (instead 9a, 9a' or 9') for this compound. About the configuration of the stereogenic centers, they were established by the energetic calculation [20] of the isomeric structures shown in scheme 4 (9, 9' and 9") being 9 those having the lesser energy (Scheme 4).
Additional spectroscopic features of compound 9 also support such structure. For example the MS of the oxidized derivative 9 shows a molecular ion at m/z 416 (FAB). The 1H NMR shown the usual doublets for the vinylic protons at δ = 6.97 and 6.22 (J = 16.0 Hz) and the corresponding methyl singlets at δ = 2.22 (3H), 1.08 (6H) and 0.86 (3H). The 13C 1D and DEPT edited spectra allowed the partial assignment of the chemical shift of the remaining carbon atoms (protonated carbons) at δ = 35.4, 33.5, 29.7, 28.1, 25.8 (2C), 20.8 and 16.8. The singlets at δ = 197.5 (C-12), 70.5 (C-2), 65.8 (C-1) and 33.5 (C-3) matching the spectrum for such a structure. The tentative approach of the mechanism involved in the above mentioned dimerization is displayed in scheme 3.
On the other hand, the new furenone 10 displayed at IR spectrum an absorption at 1762 cm−1, which clearly indicates an α, β unsaturated γ-lactone moiety. The absorbance at UV spectrum λmax = 295 nm, reflects an α, β, γ cromophore [21]. The proton spectrum showed a one proton quartet at δ = 7.13 4J = 1.2 Hz and a doublet methyl group at δ = 2.05. The former was converted to a singlet when the methyl protons at δ = 2.05 was decoupled. The 300 MHz 1H NMR showed in addition methyl singlets at δ = 1.58 (C-7), 1.30 (C-8) and 1.29 (C-9). Both, the strong IR band at 1762 cm−1 and the 13CNMR singlet at δ = 170.4 indicate the presence of an enol lactone carbonyl. The DEPT edited spectrum allowed the observation of two methines at δ = 137.8 (C-11) and 36.2 (C-6). Also, two methylene carbons at δ = 37.7 and 17.3 assigned for C-4 and C-5 respectively. Finally, four methyl signals at δ = 10.7, 29.2, 28.1 and 29.6 for C-13, C-7, C-9 and C-8 respectively. The 1D NMR enabled us the assignment of the nonprotonated carbons as δ = 149.1 (C-10), 128.3 (C-12), 106.1 (C-2), 71.2 (C-14), 35.0 (C-3), and 29.6 (C-1).
A new unexpected trichloro derivative 11 whose structure emerge from its observed MS molecular ions [m/z (M+) 309, (M + 2) m/z 311, (M + 4) m/z 313, and (M + 6) m/z 316]. The proton NMR spectrum showed a one proton doublet of doublet δ = 7.12 (J = 15.5, 10.5 Hz) and an one proton doublet of doublet at δ = 6.41 (J = 0.5, 15.5) for CH-10 and CH-11 respectively. A new methylene signal appeared at δ = 4.22 instead of the usual methyl ketone singlet (δ = 2.31) observed in the 1HNMR spectrum of starting β-ionone 1 (Fig. 3).
Additionally, we were able to observe a doublet of doublet at δ = 2.40 (J = 10.5, 0.5, CH-2) and three methyl singlets at δ = 1.30, 0.95 and 0.78 for C-7, C-8, and C-9 respectively.
The 1D 13C NMR and DEPT edited spectra allowed the assignment of protonated carbons as four methines at δ = 148.0 (C-10), 129.1 (C-11), 48.1 (C-2) and 32.8 (C-6) respectively. Three methylenes at δ = 47.2, (C-13), 35.1 (C-4) and 15.9 (C-5). Three methyl signals were also observed at δ = 29.4, 22.5, 19.7 for C-8, C-7 and C-9 respectively. The assignment of the non protonated carbons at δ = 190.5 (C-12), 72.8 (C-14), 32.0 (C-3) and 29.8 (C-1) was performed using the COLOC [18] spectrum.
The formation of compounds 10 and 11 may be inferred through isomerization [22-26] of the bond C6-C1 from compound 1' to 1 respectively and then the dichlorocarbene addition to the new formed double bond.
On the other hand, the role of the catalyst used in this work, play an important role in the ratio of the products obtained as well as the versatility of the structures and yield. Graphics 1 and 2 are self explained and show the behaviour of the different catalyst and ratio of products.
Conclusions
The successful use of several ammonium quaternary catalyst in the phase transfer reactions enabled us to obtain some interesting derivatives from the starting material (β-Ionone). The structures of the formed compounds where fully elucidated using 2DNMR, X-ray crystallography, chemical shifts and molecular modeling calculations.
Experimental
Melting points were determined with a Kofler Hot Stage apparatus and were not corrected. The NMR 1H and 13C spectra were recorded using Varian Unity 300 spectrometer operating at observation frequency of 300.0 MHz for 1H and 75.0 MHz for 13C. The 1H and 13C chemical shifts (δ) are given in ppm relative to tetramethyl silane (TMS). The COSY, NOESY, HETCOR, DEPT and COLOC spectra were recorded using the usual Varian Unity software.
High resolution spectra were recorded on a Varian Unity 500 operating at 500.3 MHz for 1H and 125.0 MHz for 13C. The experiments were performed using an inverse detection 5 mm probe. The COSY, NOESY, HMQC and HMBC experiments were performed using the usual Varian Unity software.
Mass spectra were recorded on instruments using CI/EI sources on a JEOL-JMS-AX505 HA and JEOL-JMS-10217. The IR spectra were performed on Nicolet FX-sx and Nicolet 55-X in film mode.
The β-ionona was purchased from Aldrich Chemical and used as received. The ammonium quaternary catalyst were kindly provided by Akzo Chemical Chicago Ill. and they received the trade name usually used. Benzalconium Chloride (1); Arquad HT-50 (II); Arquad 2C-75 (III); Arquad S-50 (IV); Arquad 316 (V); Ethoquad C-12 CB75 (VI); Propoquad C-12 O2A (VII); Arquad M2H TB80 (VIII); Arquad 16-25W (IX); Propoquad T-12 O2A (X); Tetramethyl ammonium acetate (XI); Arquad 2HT-75 (XII); Ethoquad O-12 (XIII); Arquad 12/50 (XIV); Arquad 16/29 (XV); Arquad T-50 (XVI); Arquad 2HT50 (XVII); Arquad 16/50 (XVIII); TEBAC (XIX); Dimethyldidecyl ammonium chloride (XX); Tetrabutyl ammonium chloride (XXI).
2,6,6-trimethyl-1-(2-acetyl-3,3-dichlorocyclopropyl) cyclohexene 4. Colorless crystals, mp 54-56 °C. C14H20OCl2. M.W. 274. 1HNMR. δ 0.96 2 (CH3-8); 1.01 s (CH3-9; 1.37 m (CH2-4); 1.60 m (CH2-5); 1.75 s (CH3-7); 2.05 m (CH2-6); 2.44 s (CH3-13); 2.86 (CH-10), 3J = 9.0, 5J = 1.3; 3.10 d J = 9 (CH-11). 13C NMR. δ 196.3 O=C-12; 136.1, C-2; 128.6, C-1; 64.4, C-14; 44.1 ,C-11; 41.2, CH2-4; 38.3, C-10; 34.5, C-3; 32.8, CH2-6; 32.1, CH3-13; 28.7, CH3-8; 28.6, CH3-9; 21.1, CH3-7; 19.5, CH2-5. IR νmax cm−1 2967, 2934, 2869, 1718, 1170, 842. MS EI m/z M+ 274 (13); M++2, 276 (7); M++4, 278 (3); 231 (87); 195 (49); 43 (100).
7,7-dichloro-2,5,6-trimethyl-1-(3-oxo-1-butenyl)bicyclo[4,1,0] heptane. 5. Colorless liquid C14H20OCl2 MW 274. 1HNMR δ 1.01, s CH3-9; 1.22, s CH3-8; 1.23, s CH3-7; 1.42, m CH2-4; 1.84, m CH2-5; 2.02, m CH2-6; 2.28, s CH3-13; 6.16 d, CH-11 3J = 16.5; 6.63, d CH-10 3J = 16.5. 13C NMR δ. 197.7 O=C-12; 143.2, C-10; 136.1, C-11; 76.3, C-14; 40.9, C-3; 35.9, C-4; 33.8, C-2; 32.1, C-1; 30.2, CH3-8; 28.2, C-6; 27.4, CH3-13; 25.7, CH3-7; 22.7, CH3-9; 17.9, C-5. IR νmax cm−1; 2959, 2935, 2870, 1678, 1253, 837. MS. EI m/z M+ 274 (3); M++2, 276 (1); M++4, 278; 259 (27), 196 (38); 181 (88); 161 (53); 123 (100); 43 (97).
2-chloro-5-methyl-3-(2,6,6-trimethyl-1-cyclohexenyl) furane. 6. Colorless liquid. C14H19OCl, MW 238 1HNMR. δ 0.91, s CH3-8; 1.00, s CH3-9; 1.43, s CH3-7; 1.53, m CH2-4; 1.77, m CH2-5; 2.04, m CH2-6; 2.25, d , 4J = 1.0, CH3-13; 5.80, q, 4J = 1.0, CH-11. 13C NMR δ 149.9, C-12; 132, C-2; 131.1, C-14; 130.1, C-1; 119.5, C-10; 111.0, C-11; 39.1, C-4; 34.9, C-3; 32.0, C-6; 29.1, C-8; 28.3, C-9; 20.8, C-7; 19.3, C-5; 13.6, C-13. IR νmax cm−1. 2960, 2931, 2867, 2832, 1614, 1236. MS EI m/z M+ 238 (38), M++2, 240 (15); 223, (100); 187 (34); 159, (24); 145, (31); 129, (53); 115, (40), 91, (50); 77, (49); 65, (35).
4-[4,4,4b, 8,8, 10a-hexamethyl-4a (3-oxo-but-1-enyl)-decahydro-9,10- dioxaphenanthren-8a-yl]-but-3-en-2-one. 9. Colorless liquid C26H40O4 MW 416. 1H NMR δppm 0.86, s CH3-9; 1.08, s CH3-8; 1.00 m, CH2-4; 1.08 s CH3-7; 1.37 m CH2-5; 1.77, CH2-6; 2.22, s CH3-13; 6.22 d 3J = 16.0 CH-11; 6.97 d 3J = 16.0 CH-10. 13C NMR δ 197.5 C-12; 142.6 C-10; 132.4 C-11; 70.5 C-2,65.8 C-1; 35.4 C-4; 33.5 C-3; 29.7 C-6; 28.1 CH3-13; 25.8 (2C) CH3-8, CH3-9; 20.8 CH3-7; 16.8 C-5. IR νmax cm−1 2960, 2935, 2872, 1677, 1627, 1255. MS FAB m/z M+ 416, 400. 383, 355, 341, 327, 209, 191, 123(100), 69, 43.
5-(7,7-dichloro-1,3,3-trimethylbicyclo[4,1,0]heptyl-2-iden)-3-methyl-Furan-2-one. 10. Colorless solid m. p. 80-81 °C C15H18Cl2O2 MW 300. UV λmaxnm 295. 1H NMR δppm 1.29, s CH3-9; 1.30, s CH3-8; 1.43, m CH2-4; 1.58, s CH3-7; 1.68, q CH2-6; 2.10, m CH2-5; 2.05, q CH3-13; 7.13, q H-11. 13C NMR δ 170.4 C-15; 149.1 C-10; 137.8 C-11; 128.3 C-12; 106.1 C-2; 71.2 C-14; 37.7 C-4; 36.2 C-6; 35.0 C-3; 29.6 C-8; 29.2 CH3-7; 28.1 CH3-9; 27.2 CH3-7; 17.3 C-5; 10.7 CH3-13. IR νmax cm−1 2932, 2867, 1762,1207, 802, 741. MS EI, m/z M+ 300 (51), M++ 2, 302 (35); M++ 4 304 (7); 285, 265, 244, 217, 169 (100); 142 (78), 138 (60).
7,7-dichloro-1,3,3-trimethyl-2-(3-oxo-4-chloro-1-butenyl)bicyclo[4,1,0] heptane. 11. 1H NMR δppm. 0.78 (s) CH3-9; 0.95 (s) CH3-8; 1.30 (s) CH3-7; 1.23, (m) CH2-4; 1.40 dd, J=10.0, 8.0, CH-6; 1.93 (m) CH2-5; 2.38 (m) CH-2; 4.22 CH2-13; 6.41, dd J=0.5, 15.5, CH-11; 7.12, dd J=10.5, 15.5, CH-10. 13C NMR δ 190.5 (C-12), 148.0, (C-10); 129.1 (CH-11); 72.8 (C-14); 48.1 (CH-2); 47.2 (CH2-13); 37.8 (CH-6); 35.1 (CH2-4); 32.1 (C-3); 29.8 (C-1); 29.4 (CH3-8); 22.5 (CH3-7); 19.7 (CH3-9); 15.9 (CH2-5). MS CI M+ m/z 309 (51), M + 2 m/z 311 (42); M + 4 m/z 313 (17); M + 6 m/z 316 (6); 273 (86); 237 (70); 217 (80); 169 (100); 123 (58). IR νmax cm−1. 2957, 2930, 2867, 1715, 1697, 838.
Acknowledgements
We thank M. I. Chávez and B. Quiroz for the NMR determinations. We also thank R. Patiño, L. Velasco and R. A. Toscano for the IR, MS and X-ray determinations, respectively. L. Muciño thanks to CGI y EA of UAEM and SNI-Conacyt for partial financial support. We thank also E. Rivera of Akzo Chem. Chicago Ill. For the samples of ammonium quaternary catalyst used in this work.
References
1. a) Aasen, A. J.; Kimland, B.; Almquist, S.D.; Enzell, C.R. Acta Chem. Scand. 1972, 26, 2573-2576. [ Links ] b) Wahlberg, I.; Enzell, C. R. Nat. Prod. Rep. 1987, 4, 237-276. [ Links ]
2. Kernan, M. R.; Faulkner, D. J.; Jacobs, R. S. J. Org. Chem. 1987, 52, 3081-3083. [ Links ]
3. Demole, E.; Enggest, P.; Winter, M.; Furrer, A.; Sculte-Elte, K. H.; Egger B.; Ohloff, G.; Helv. Chem. Acta 1979, 62, 67-75. [ Links ]
4. Behr, D.; Wahlemberg, I.; Nishida, T.; Enzell, C. R. Acta Chem. Scand. Ser. B 1977, B31, 609-613. [ Links ]
5. a) Buchi, G.; Vederas, J. C. J. Amer. Chem. Soc. 1972, 94, 9128-9132. [ Links ] b) Demole, E.; Berthet, D. Helv. Chim. Acta 1981, 54, 681-686. [ Links ] c) Snowden, R. L.; Linder, S. M.; Muller, M.L.; Shulte-Elte, K .H. Helv. Chim. Acta 1987, 70, 1858-1878. [ Links ]
6. Findlay, J. A.; MacKay, W.D. Can. J. Chem. 1971, 49, 2369-2371. [ Links ]
7. a) Roberts, D. L.; Heckman, R. A.; Hege, B. P.; Bellin, S. A. J. Org. Chem. 1968, 33, 3566-3569. [ Links ] b) Ontani, T.; Yamashita, K. Tetrahedron Lett. 1972, 2521-2524. [ Links ]
8. Demlov, E. V.; Prashad, M. J. Chem. Res. 1982, 354. [ Links ]
9. Baird, M. S., Baxter, A. G. W.; Devling, B. R. J.; Searle, R. J .G. J. Chem. Soc. Chem. Commun. 1979, 210-211. [ Links ]
10. Sydnes, L. K. Acta Chem. Scand. Ser B 1977, 31, 823-828. [ Links ]
11. Sydnes, L. K.; Skattebold, L. Tetrahedron Lett. 1975, 4603-4606. [ Links ]
12. Díaz, E.; Toscano, R. A.; Alvarez A.; Shoolery, J. N.; Jankowski: Can. J. Chem. 1990, 68, 701-704. [ Links ]
13. Díaz, E.; Fuentes, A.; Villafranca, E.V.; Jankowski, K Acta Crystallographica 1994, C50, 2030-3032. [ Links ]
14. Doddrell, D. M.; Pegg, D.T.; Bendall, M. R. J. Magn. Reson. 1982, 48, 323-327. [ Links ]
15. a) Bax, A.; Subramanian, S. J. Magn. Reson. 1986, 67, 565-569. [ Links ] b) Bax, A.; Freeman, R. J. Magn. Reson. 1981, 44, 542-561. [ Links ]
16. a) Derome, A. E. in: Modern Techniques for Chemistry Research, Pergamon Press, N.Y. 1987, p 240. [ Links ] b) Martin, G. E.; Sektzer, A. S. in: Two Dimensional NMR Methods for Establishing Molecular Connectivity, VCH Publishers, N.Y. 1988, p. 219. [ Links ]
17. a) Bax, A.; Davies, D. G. J. Magn. Reson. 1985, 63, 207-213. [ Links ] b) Bax, A.; Summers, M. F. J. Amer. Chem. Soc. 1986, 108, 2093-2094. [ Links ]
18. Kessler, H.; M. Gehrke, M.; Griesinger, C. Angew. Chem. Int. Ed. Engl., 1988, 27, 490-536. [ Links ]
19. a) Furst, A.; Pretsch, E.; Robien, W. Ann. Chem. Acta 1990, 233, 213. [ Links ] b) Furst, A.; Pretsch, E. Ann. Chim. Acta 1990, 229, 17. [ Links ] c) Furst, A.; Pretsch, E.; Robien, W. Ann. Chim. Acta 1991, 248, 415. [ Links ]
20. Calculations were first minimized to 0.1 Kcalmol-1, using semi-empirical method CS Chem 3D version 5 for MacIntosh.
21. Silverstein, R. M.; Bassler, G. C.; Merril, T. C. Identificación Espectroscópica de Compuestos Orgánicos, Editorial Diana. México 1980 pp 17-85. [ Links ]
22. Wahlemberg, Enzell, C. R. Nat. Prod. Reports. 1987, 4, 237-276. [ Links ]
23. Bucherer, R.; Hamm, P.; Eugster, C. H. Helv. Chim. Acta 1974, 57, 631-656. [ Links ]
24. Molnar, P.; Szaboles, J. Acta Chim. Acad. Sci. 1979, 99, 155. [ Links ]
25. Bischofberger, N.; Frei, B.; Wirz, J. Helv. Chim. Acta. 1983, 66, 2489. [ Links ]
26. Horspool, W. M. Photochem. 1985, V16, 248. [ Links ]

