











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
Studies on extremophilic microorganisms have been a breakthrough for the fields of biochemistry (Xu and Glansdorff, 2002; Conners et al., 2006), biotechnology (Vieille and Zeikus, 2001; Guiral et al., 2012) and early evolution, broadening our understanding of the limits of life (Nisbet and Sleep, 2001; Allers and Mevarech, 2005).
Extremophiles are organisms that surpass the mesophilic limits of parameters such as temperature, pH and salinity, and can have biochemical dependencies on different organic and inorganic compounds like sulfur, nitrogen, methane or even ferrous oxides (respectively) (Stetter et al., 1990). The respiration reactions of these organisms increase our awareness of what we might find outside our planet (Trent, 2000). It is necessary to integrate a clear approach to the study of these organisms to recognize new trends and molecular signatures in different environments. One of the extremophilic lifestyles that has been heavily studied due to its impact in early evolution (Islas et al., 2003) and its biochemical diversification is the group of hyperthermophilic prokaryotes from the Archaea and Bacteria domains (Stetter, 2006). A large number of studies involving hyperthermophilic prokaryotes are based on their multiple strategies for survival and their stability in high-temperature environments (Stetter et al., 1990). There is no single molecule or metabolic pathway unique for this lifestyle (Atomi et al., 2004), and finding a pattern or common properties in all the described species remains elusive (Trent, 2000; Allers and Mevarech, 2005). However, it is possible to associate some molecular traits with nucleotide and amino acid levels, beyond any phylogenetic groups with hyperthermophilic character. In this regard, Groussin and Gouy (2011) recognize two main characteristic processes in the hyperthermophilic lifestyle: a) the molecular evolutionary rate is skewed by the optimal growth temperature and tRNA-coding genes, and b) it is possible to correlate the optimal growth temperature with multiple components of coding and non-coding sequences. Also, Klipcan et al. (2006) correlated the optimal growth temperature with the proportion and type of aminoacyl-tRNA synthases (aatRNAs). Agarwal and Grover (2008) recognize a purine bias that modifies the amino acid frequency and codon usage. Therefore, it has not yet been possible to recognize common genes for all the hyperthermophilic genomes, but it is feasible to discover biases and proportions in amino acids and nucleotides that could define a differential composition in this lifestyle. As it has been proposed (Cordero and Polz, 2014), the hyperthermophilic genome can be studied in two main sections: 1) the core genome that is composed of all the housekeeping genes involved in basic metabolism, and 2) the flexible genome, which is shaped by genes related to habitat-specific properties, as well as interaction between viruses and predators. These genes have a variable presence in the entire genome, and are involved in horizontal gene transfer events, gene loss and high rates of gene turnovers.
With these pangenomic studies, it is possible to infer that the bacterial chromosomes are composed of genes from the core and flexible genomes, but the extrachromosomal materials, like the plasmids, are composed mainly of genes related with the flexible genome.
By studying the plasmid genome, it is possible to recognize the arrangement of the structure of the DNA and the variance of codon usage in the same organism. Berg and Kurland (2002) and Cordero and Polz (2014) proposed that the study of the flexible genome allows the identification of global or individual genes that can be used as the reflection of the prokaryotic community sharing the same environmental conditions. Also, according to Cooper et al. (2010), secondary bioinformational elements (like secondary chromosomes, megaplasmids, or plasmids) show a decrease in codon usage diversity in the case of organisms living in the same environment, despite belonging to different phylogenetic groups.
Since codon usage have a differential appearance in the coding sequences, there are different ways to measure the structure and topology of the genome. One approach consists of recognizing dinucleotide interaction in the entire DNA sequence and correlating it with the DNA twist profile. As noted by Quintana et al. (1992), the DNA crystallography profiles correlate the DNA twist with the space and configuration among dimers. The High twist profile (H value) indicates the incidence of sequence of dinucleotides GC or GA, and elevated values of DNA twist and space among nucleotide dimers. In contrast, the Low twist profile (L value) is present where the sequence has a high frequency of CC, CT, TT, AA, AG and GG dimers. These configurations present the values of twist and separation between the dimers. The Variable twist profile (V value) is a combination of both configurations and is correlated with the incidence of pyrimidine and purine dimers, a conformation that is strongly susceptible to the influence of the environment.
This paper focuses primarily on the comparative genomics, with special emphasis on the evolution of the extremophilic genomes. For the codon usage analysis, we review Jaenicke et al. (1991), which implied that a decrease in non-charged amino acids allows discrimination between extremophile and mesophile proteomes. Also, Zeldovich et al. (2007) correlate the increase in the use of seven amino acids (IVYWREL) with a raise in the optimal growth temperature. Using both approaches-analysis in DNA twist profile and codon usage-we tried to identify a shared trait among the chromosomes and plasmids of some hyperthermophilic organisms. If found, we might recognize common characteristics for the hyperthermophilic lifestyle.
2. Methods
In order to classify and recognize the optimal temperature interval for the proposed archaeal and bacterial species, we use the National Center for Biotechnology and Information (NCBI) database (http://www.ncbi.nlm.nih.gov). These data are compiled in Table 1. A total of eight hyperthermophilic species with complete chromosome and plasmid sequences were obtained from the ftp site of NCBI. The eight species include three Crenarchaeota (Sulfolobus islandicus L.D.8.5, Sulfolobus islandicus Y.N.15.51, and Thermofilum pendens Hrk5), four Euryarchaeota (Archaeoglobus profundus DSM 5631, Methanococcus maripaludis C5, Pyrococcus abyssi and Thermococcus barophilus MP), and one species from Bacteria (Aquifex aeolicus VF5). A mesophilic organism (Escherichia coli O157 H7) was used as a negative control organism. We chose this strain because of their mesophilic lifestyle and the incidence of their unique plasmid structure and representation. The H, V and L mean values were evaluated on genomes and plasmids using the program codon.pl.
Table 1 Thermophilic and hyperthermophilic genomes of the Archaea and Bacteria domains used in this paper.
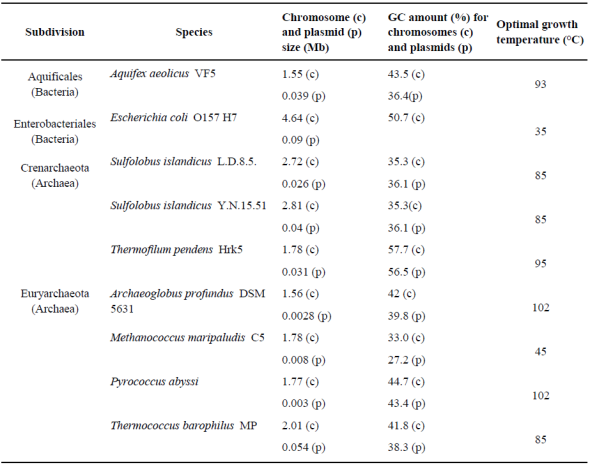
Note. The data from the chromosomes (c) and plasmids (p) come from the NCBI database, and the characteristics and the optimal growth temperature from Horneck and Baumstark-Khan (2002).
Additionally, using the UGENE 1,19 software (Okonechnikov et al., 2012), we calculated high flexibility areas and tandem repeat sequences in both chromosomes and plasmids. The module that would recognize the high flexible sequences was applied using the default values, and the tandem repeat module was modified to recognize sequences greater than 20 nucleotides. The size of tandem repeats is based on previous reports (van der Oost et al., 2014).
In this study, we selected only completely sequenced genomes of organisms with reported thermophilic or hyperthermophilic lifestyles (Stetter et al., 1990; Horneck and Baumstark-Khan, 2002), separated into chromosome and plasmid. Also, the genome of Methanococcus maripaludis was included because of their thermophilic tolerance, and it would be important to have a comparative example with a methanobacterial genome.
3. Results
3.1. Genome sampling in the NCBI database
The information in Table 1 shows the available diversity of archaeal and bacterial hyperthermophilic species, with complete chromosome and plasmid sequences. We analyzed eight species: one from the bacterial domain, three from Crenarchaeaota subdomain, and four from the Euryarchaeota subdomain. The comparison of the GC amount in the plasmids and chromosomes shows that this value in Crenarcheota is more stable than in the other groups. In addition, two species with an increased genome size were found. Crenarchaeota has a stable genomic structure, unlike the Euryarchaeota that has differences among the plasmid and chromosome GC values.
3.2. V, H and L comparative values
In Figure 1, the three main profiles of DNA twisting are integrated. The results cluster into the same area in which the H value increases in diverse chromosomes and plasmids, and the L value is positive and contains ~ 75 % of the sample. The only exceptions to this grouping are the Thermofilum pendens genome, the genome and plasmid of Escherichia coli O157 H7 and the Methanococcus maripaludis plasmid. Additionally, we identified a large difference in the V values for the chromosome and plasmid of Aquifex aeolicus and the plasmid of Archaeoglobus profundus. This does not correspond with a phylogenetic signal, or a similar optimal growth temperature or even GC amount. The result was similar for Aquifex and Archaeoglobus plasmids that show overlapping and similar values of twisting.
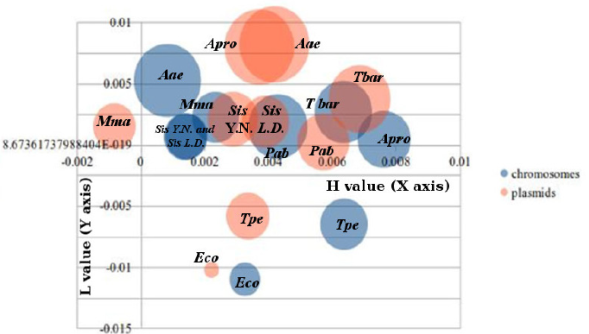
Figure 1 Graphical representation of the profile values for the chromosomes and plasmids of hyperthermophiles. The H and L values were used for abscises and ordinates, respectively, and the V value were used as an absolute number for bubble size representation. All values in %. The name of the species are pointed as follows: Aae = Aquifex aeolicus VF5; Sis = Sulfolobus islandicus (L.D. = L.D.8.5.; Y.N. = Y.N.15.51); Tpe = Thermofilum pendens Hrk5; Apro = Archaeoglobus profundus DSM 5631; Mma = Methanococcus maripaludis C5; Pab = Pyrococcus abyssi and Tbar = Thermococcus barophilus MP; Eco = Escherichia coli O157 H7. Obtained values for chromosomes are in blue, and for plasmids in red.
3.3. Tandem repeat sequences and High flexibility regions
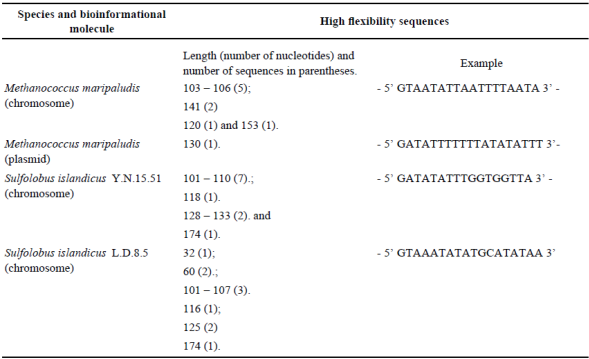
In order to recognize if all the plasmids show the same flexibility in their genome, or if they share a common aspect in this feature, we analyzed the incidence of particular regions and coupled it with tandem repeat regions (Allers and Mevarech, 2005; Norais et al., 2013). The results are shown in Table 2 and 3. Contrary to previous models, high flexibility values for plasmids were not recognized. Only the plasmid of M. maripaludis has high flexibility and shares a similar L value with the chromosome. The structure of the chromosome allows both high flexibility regions and tandem repeat regions. The chromosome of both Sulfolobus species shows both elements. This signal is not shared by any of the analyzed hyperthermophilic genomes.
Table 2 Tandem repeat sequences identified in genomic hyperthermophiles.

Note. Only sequences and regions with the default values of UGENE are recognized.
3.4. Codon usage and amino acid values
For the correlating values, we notice that the highest values occur among plasmids (Figure 2), with R2 > 0.7 for leucine and isoleucine, whereas the highest value for chromosomes (Figure 3) is shown by glutamic acid (R2 ≈ 0.7).
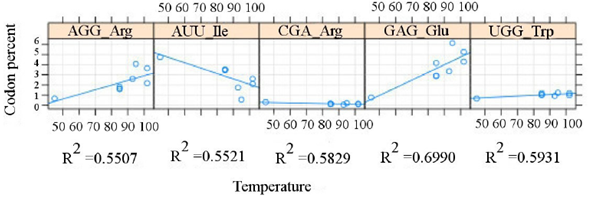
Figure 2 Correlation between the codon usage in chromosomes and the optimal growth temperature (OGT). The relation between the reported optimal growth temperature (abscises) versus the percent value (ordinates), as evaluated from the overall diversity of the genetic code, is presented. The R2 value of each resulting codon is shown below the corresponding area.
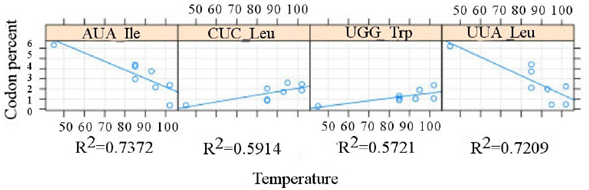
Figure 3 Correlation between the codon usage in plasmids and the optimal growth temperature (OGT). The relation between the reported optimal growth temperature (abscises) versus the percent value (ordinates), as evaluated from the overall diversity of the genetic code, is presented. The R2 value of each resulting codon is shown below the corresponding area.
4. Discussion
4.1. Genome sampling and further samples
Although the sample used is relatively small, important conclusions can be drawn. This work provides an approach as to how the hyperthermophilic Bacteria and Archaea can be arranged for DNA twisting in chromosomes and plasmids. One case that needs further study is the arrangement of Sulfolobales, where both sets of analyzed chromosomes have an identical DNA twist value (for H and L values), and a different value for their plasmids (implied in the H value). The Sulfolobales order still needs further study, including different species like S. acidocaldarius to confirm their structure and variation as a phylogenetic trend that could complement the previously reported high flexibility genomes (Zillig et al., 1996; Farkas et al., 2011).
Increasing the amount of complete hyperthermophilic genomes, especially bacterial, will provide a better understanding of the flexible genome perspective.
4.2. H, L and V values, and their correlation with the hyperthermophilic lifestyle
Although not all values are correlated directly with the hyperthermophilic lifestyle, we can recognize similar V and L values; this applies to 75 % of the sample. This proportion occurs in both chromosomes and plasmids, and may imply that the amount related to Low twist profile (L) arrangement and Variable twist profile (V) in the archaeal hyperthermophilic genome is a main trend.
The eccentric position of the T. pendens genome could be explained by the high incidence of events of gene loss (Anderson et al., 2008), and the effect of a recent split transfer genes involving informational genes (Chan et al., 2011). M. maripaludis shows a negative H value, and it is possible that the DNA twist and structure are associated with its thermotolerant and not hyperthermophilic lifestyle. Furthermore, it has been reported that the Methanobacteriales have a high incidence of gene conversion (Hildenbrand et al., 2011) that could modify their base composition and gene arrangements. After integrating Escherichia coli O157 H7 as a mesophilic control, its proportion and incidence, graphed in a different area of Figure 1, provides a good comparison with mesophilic values.
The nearness of the V and H values of plasmids from Archaeoglobus profundus and Aquifex aeolicus could be evidence of the high horizontal gene transfer events between them (van Wolferen et al., 2013). However, it is necessary to develop further pangenomic analysis and comparative studies of these sequences.
4.3. Tandem repeats and High flexibility sequences
The highest incidence of tandem repeats and high flexibility sequences in chromosomes allowed us to identify punctual regions involved in recombination and increased flexibility and possibly "hotspots" of mutation and recombination.
The finding of only one sequence with repeats in the plasmid of M. maripaludis suggests that plasmids from hyperthermophilic species need additional analyses. This result contrasts with the general idea in which tandem repeats and high flexibility sequences are correlated with recombination events (Johnson et al., 2013) and with the occurrence of DNA repair mechanisms (Cai et al., 2009) that shift the structure of the plasmid DNA. Moreover, this suggests that plasmids in hyperthermophilic organisms might be regulated by different mechanisms in their chromosomes.
4.4. Codon usage and amino acid proportion
By comparing the codon usage for the seven proposed amino acids that correlate with thermotolerance, we have a different pattern than previously published by Zeldovich (Zeldovich et al., 2007). Although it is impossible to associate the increase of certain codon amino acids to the role of thermal stabilizers, this bring us an additional point of view about the genome structure response to the extremophilic lifestyle.
The proposed analysis allowed the recognition of different patterns in the same amino acid. An example of this differential pattern is the decreasing isoleucine usage, which is negatively correlated for chromosome and plasmid for all the analyzed hyperthermophiles, although with different codons. On the other hand, leucine is positively correlated with OGT (codon CUC) and negatively in plasmids (with codon UUA). This disagrees with Zeldovich et al. (2007), and consequently a more detailed analysis is required. That would include other extremophilic groups.
Furthermore, the increase in glutamic acid is significant in chromosomes along with an increase in OGT, while in the plasmid the UGG codon (tryptophan) increases with OGT. It has been proposed that glutamic acid is relevant for hyperthermophilic organisms because of its charge, thus causing a difference in side chain entropy, and helping in protein folding under extreme conditions (Greaves and Warwicker, 2007). However, tryptophan is an amino acid whose amount decreases into thermostable proteins (de Champdoré et al., 2007), and shows a small increase in plasmid sequences.
With this, we infer that proteins coded in the chromosomes show different performances from those coded in the plasmid. This bias in the plasmid could be explained by their accessory role in the metabolism of hyperthermophilic organisms.
5. Conclusions
The study of the dynamic DNA structure of archaeal and bacterial hyperthermophiles allows further understanding of the biology of this particular lifestyle. We determined that the H, V and L values are similar for all the analyzed organisms.
There is not satisfactory correlation on the changing trend of codon usage in chromosomes and plasmids with skews in the coding sequence.

