










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.82 no.2 Ciudad de México abr./jun. 2012
Artículo de revisión
Principios básicos de las nuevas estrategias farmacológicas antiarrítmicas en fibrilación auricular
Basic mechanisms of the new antiarrhythmic drugs in atrial fibrillation
David Filgueiras–Rama,1,2 Sergio Castrejón,1 Conrado Calvo,2 Alejandro Estrada,1 David Doiny,1 Marta Ortega,1 Omer Berenfeld,2 José L. Merino,1 José Jalife.2
1 Unidad de Arritmias y Electrofisiología Cardiaca Robotizada. Departamento de Cardiología. Hospital Universitario La Paz. Madrid, España.
2 Centro para Investigación de Arritmias. Departamento de Medicina Interna, Medicina cardiovascular. Universidad de Michigan. E.U.
Correspondencia:
Dr. David Filgueiras Rama.
Unidad de Arritmias y Electrofisiología Cardiaca Robotizada.
Hospital General La Paz. Paseo de la Castellana 261. C.P. 28046. Madrid, España.
Teléfono: (+34) 91 727 7564. Fax: (+34) 91 727 7564.
Correo electrónico: david.filgueiras@salud.madrid.org
Recibido el 22 de septiembre de 2011.
Aceptado el 6 de diciembre de 2011.
Resumen
La fibrilación auricular (FA) es la arritmia crónica sostenida más frecuente en la población general. A pesar de los últimos avances tecnológicos y en el entendimiento de sus mecanismos, derivados de modelos experimentales, así como de los procedimientos de ablación en pacientes con FA, los fármacos antiarrítmicos siguen siendo la principal estrategia para la cardioversión y mantenimiento del ritmo sinusal. Nuevas generaciones de fármacos antiarrítmicos han llegado a la práctica clínica, y otros se encuentran en fase de experimentación. Los nuevos fármacos actúan de forma más específica sobre corrientes iónicas auriculares, y al mismo tiempo involucradas en el mantenimiento de la arritmia. Paralelamente, cada vez se da más importancia a la necesidad de actuar sobre el sustrato arritmogénico auricular y los factores que lo promueven, implicados en el mantenimiento a largo plazo de la arritmia (terapias upstream). La presente revisión tiene como objetivo exponer las actuales líneas de desarrollo en fármacos antiarrítmicos y terapias para prevención o retraso del remodelado auricular, con base a los conocimientos mecanísticos que hoy en día se involucran en el mantenimiento de la FA.
Palabras clave: Fibrilación auricular; Rotores; Fármacos antiarrítmicos; Corrientes iónicas; Remodelado auricular; Dronedarona; España.
Abstract
Atrial fibrillation (AF) is the most common sustained arrhythmia seen in clinical practice. Despite of new technological breakthroughs and the understanding of the mechanisms underlying AF, based on animal models and ablation procedures in patients, the antiarrhythmic drugs remain the main therapeutic strategy to restore and maintain the sinus rhythm. New antiarrhythmic drugs are already available in the clinical practice and many others are under development. The new antiarrhythmic drugs have the capability to block atrial–specific ionic currents, which are involved in the maintenance of the arrhythmia. Parallel, increasing evidence supports the use of compounds to regulate the arrhythmogenic atrial substrate involved in the long–term maintenance of the arrhythmia (upstream therapies). This article reviews the new antiarrhythmic drugs and upstream therapies, based on the current knowledge of the mechanisms involved in the maintenance of AF.
Keywords: Atrial fibrillation; Rotors; Antiarrhythmic drugs; Ion currents; Atrial remodeling; Dronedarone; Spain.
Introducción
La fibrilación auricular (FA) es la arritmia crónica sostenida más frecuente en la población general.1 Actualmente, se estima que en EEUU unos 2.5 millones de personas padecen FA y lo que es más importante, estudios poblacionales pronostican que su prevalencia se incrementará de forma significativa en los próximos años, pudiendo triplicarse en el año 2050.2 La FA dobla el riesgo de mortalidad en ambos sexos, y supone un factor de riesgo mayor para ictus embólico.3 Los pacientes con FA también presentan deterioro en su calidad de vida, lo cual en muchos casos, representa el principal motivo por el que se decide una estrategia de control de ritmo.
A pesar de los últimos avances tecnológicos y en el entendimiento de los mecanismos derivados de modelos experimentales, así como en los procedimientos de ablación,4 los fármacos antiarrítmicos siguen siendo la principal estrategia para la cardioversión y mantenimiento del ritmo sinusal (RS). Sin embargo, el uso de fármacos se asocia a efectos secundarios sustanciales y a proarritmia.5 Recientemente, nuevas generaciones de fármacos antiarrítmicos han llegado a la práctica clínica y otros se encuentran en fase de experimentación. En líneas generales, los nuevos fármacos actúan de forma más específica en corrientes iónicas auriculares y al mismo tiempo involucradas en el mantenimiento de la FA, evitando de esa manera efectos proarrítmicos potencialmente graves como las taquicardias ventriculares. Por otro lado, se busca el desarrollo de nuevas sustancias con perfil de bloqueo multicanal en base a fármacos de eficacia demostrada, pero con menor toxicidad y similar o superior eficacia. Sin embargo, los nuevos antiarrítmicos presentan el potencial riesgo de aparición de nuevos efectos secundarios.6 Otras sustancias con estrategias más innovadoras, como el control de la homeostasis del Ca2+ intracelular y actuación sobre conexinas y uniones abiertas están empezando a ser estudiadas (Figura 1).
Cada vez se da más importancia a la necesidad de actuar sobre el sustrato arritmogénico auricular, y sobre los factores que promueven el mantenimiento a largo plazo de la arritmia. Nuevos compuestos y nuevas indicaciones de fármacos ya disponibles tratan de controlar el sustrato arritmogénico auricular (fibrosis, hipertrofia ventricular, inflamación, etcétera), en lo que se conoce como terapia upstream.
La presente revisión tiene como objetivo exponer las actuales líneas de desarrollo en fármacos antiarrítmicos y terapias para prevención o retraso del remodelado auricular, con base en los conocimientos mecanísticos, que hoy en día se involucran en el mantenimiento de la FA.
Métodos
Se realizó una revisión sistemática de la literatura médica con el objetivo de exponer una visión crítica y actual de los principios básicos de los nuevos fármacos antiarrítmicos en FA. Las bases de datos Pubmed y Medline fueron utilizadas para la búsqueda de artículos usando combinaciones de los términos "fibrilación auricular", "fármacos antiarrítmicos", "rotores", "corrientes iónicas", "mecanismos iónicos", "dinámica de ondas", "mapeo", "remodelado auricular" y "terapias upstream". Los artículos fueron seleccionados en función de su impacto científico en inglés y español, así como por su relevancia clínica en el entendimiento y manejo de la FA. La lista de referencias de cada artículo fue revisada para la identificación adicional de publicaciones relevantes.
Mecanismos involucrados en el mantenimiento de la FA
En los últimos 100 años, se ha intentado entender el mecanismo fundamental por el cual la FA se mantiene a lo largo del tiempo. De forma consistente en todos los modelos propuestos,7,8 la coexistencia de un sustrato heterogéneo y de zonas de conducción lenta favorecen la aparición de reentrada y FA. A partir de los años 50's, se estableció con gran fuerza la teoría de Moe y Abildskov,9,10 según la cual la FA puede perpetuarse en el tiempo por la presencia de múltiples frentes de onda independientes, que se distribuyen de forma aleatorizada, en presencia de un sustrato heterogéneo con baja velocidad de conducción (VC) y una masa auricular suficiente. Aunque esta teoría se mantiene en la actualidad, los nuevos avances en mapeo de alta resolución y modelos computacionales han hecho que de forma cada vez más generalizada, se acepte que la presencia de un único o pequeño número de fuentes de reentrada funcional (rotores), localizados preferentemente en venas pulmonares y pared posterior de aurícula izquierda,11 pueden mantener la FA durante periodos prolongados de tiempo.12–14 El entendimiento de la reentrada en estos casos requiere introducir el concepto de que los obstáculos alrededor de los cuales, la actividad reentrante se sostiene, pueden ser funcionales, sin la presencia de un obstáculo anatómico fijo.15 Bajo ciertas condiciones de excitabilidad, la presencia de un obstáculo anatómico o funcional con bordes marcados puede desestabilizar la propagación de las ondas eléctricas, dando lugar a la formación de actividad reentrante en forma de espiral (Figura 2C, D, E y F).16 En el punto en el que se produce la fragmentación del frente de propagación, la curvatura alcanza su máxima angulación, lo cual impide que el frente despolarizador sea capaz de invadir y despolarizar el tejido excitable inmediatamente contiguo. Debido a esto, se genera un punto de pivote alrededor del cual el frente de propagación comenzará a rotar, adquiriendo en dos dimensiones la forma de una espiral (Figura 2A y 3A).17 En tres dimensiones (3D), el eje del rotor vendría representado por el filamento sobre el cual giraría el equivalente 3D de la espiral del rotor.18
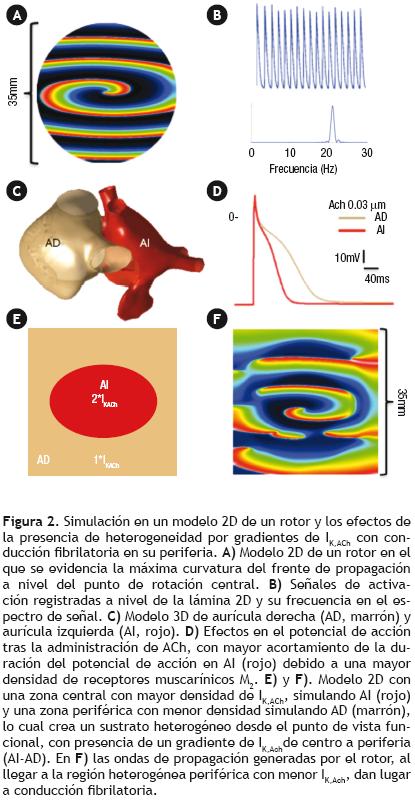
Con base en la presencia de un rotor o escaso número de rotores, que representan las fuentes de activación más rápida con valores de frecuencia dominante más altos (Figura 2A y B),19,20 la activación en la periferia se produce a través de las ondas de propagación generadas por el rotor. Los obstáculos, tanto anatómicos como funcionales, con los cuales se encuentran las ondas generadas por el rotor en su paso por las aurículas, dan lugar al fenómeno de conducción fibrilatoria (Figura 2F), que aparece representado en el electrocardiograma de superficie con las ondas de FA.21
Corrientes iónicas y rotores: su importancia en el desarrollo de nuevos fármacos antiarrítmicos
Corriente de sodio (INa)
La corriente de sodio (INa) es la principal corriente despolarizadora en el frente de onda. La reducción de la excitabilidad mediante la reducción de INa origina menor velocidad (dVdt) y amplitud en la fase 0 del potencial de acción (PA). La duración del PA (DPA) se verá acortada y la VC reducida, lo cual originará una longitud de onda (LO) menor (LO = VC x DPA).
El bloqueo de INa afectará el inicio de la reentrada, de tal manera que la reducción en excitabilidad tenderá a reducir el punto crítico de la curvatura en el que se produce el bloqueo del frente de onda de la espiral de reentrada, por la interacción con un obstáculo anatómico o funcional.22 Esto último se traduce en un circuito de reentrada con mayor longitud y mayor tamaño del núcleo central, en torno al que gira el frente de onda espiral, así como una menor LO. Por lo tanto, ante un bloqueo de INa, la reducción de la excitabilidad originará una menor probabilidad de interacción entre el frente de onda despolarizador y su cola, ya repolarizada. Esto se traduciría en una reentrada más estable y lenta. Sin embargo, el bloqueo específico de INa mediante tetrodotoxina (TTX) es capaz de detener los rotores y terminar la FA en modelos experimentales. La presencia de una reentrada más lenta podría hacer al rotor más vulnerable a la interacción y terminación, por medio de otras ondas de propagación. Asimismo, la presencia de un sustrato que fuerza al rotor a no permanecer estable, con un centro de rotación que cambia de posición, puede favorecer que el rotor choque contra bordes anatómicos y dar lugar a su terminación.23
Desde un punto de vista teórico, ambas situaciones, terminación y potenciación de la reentrada, podrían observarse en situaciones fisiológicas, dependiendo del sustrato anatómico. Está ampliamente aceptado el uso de fármacos antiarrítmicos IC para el restablecimiento del RS en pacientes con FA paroxística, con una tasa de conversión entorno al 80% en las primeras 24 horas de la administración.24 No obstante, en los casos con un sustrato más fibrótico como en FA persistente y permanente, la eficacia de los fármacos IC cae de forma drástica. Del mismo modo, la idea de un efecto proarrítmico y favorecedor de la reentrada por bloqueo de INa podría ser la causa de un aumento de la mortalidad en los pacientes con sustrato estructural ventricular después de un infarto de miocardio.25 El sustrato ventricular postinfarto asociado a un bloqueo de INa podría incrementar el riesgo de fragmentación del frente de propagación eléctrica y fomentar el inicio de reentrada.
Bloqueo selectivo de INa auricular
Se cree que las cinéticas rápidas de recuperación desde la inactivación pueden contribuir a la selectividad auricular de las sustancias bloqueadoras de Na+. De este modo, sustancias como la propafenona, que presenta una lenta disociación desde el canal de Na+ muestra poca o nula selectividad auricular, sin embargo compuestos con rápida disociación, como la ranolazina o amiodarona, tienden a ser altamente selectivos para aurículas en lo que refiere al bloqueo de INa. Cabe destacar, que la inhibición selectiva de INa obtenida hoy en día con sustancias de cinéticas de disociación rápida como la lidocaína y mexiletina, no es una estrategia muy efectiva para el manejo clínico de la FA. El bloqueo combinado de otros canales como IKr, IKur y la corriente tardía de Na+, podrían explicar el efecto altamente eficaz de sustancias como la ranolazina,26 la amiodarona y el vernakalant27 para terminar la FA.
Corriente rectificadora de entrada de potasio lNa
Aunque lNa es el principal componente de la corriente despolarizadora del frente de onda, IK1es esencial para el control de la excitabilidad cardiaca, así como la frecuencia de rotación y estabilidad de los rotores.28 IK1 tiene un papel fundamental en el control del gradiente electrotónico entre las células potencialmente excitables del núcleo del rotor y las células activas, en la periferia inmediata al núcleo. En modelos experimentales, la sobreexpresión de IK1 produce una aceleración de la fase final del PA que se traduce en una reducción en su duración, asociado a una hiperpolarización del potencial de membrana diastólico (Figura 3B). Estos cambios se traducen en menor LO durante reentrada y mayor disponibilidad de canales de Na+, lo cual incrementa la excitabilidad en el frente de onda de la espiral de rotación.29 La sobreexpresión de IK1 provocará un aumento del gradiente de voltaje entre las células activas alrededor del núcleo del rotor y las células inactivas del núcleo. Esto último contribuye al incremento de la corriente electrotónica entre las células activas y las células en reposo del núcleo, lo cual se traduce en mayor aceleración de la repolarización de las células activas y reducción de la VC en la vecindad del núcleo del rotor.29 Estos cambios provocan la presencia de rotores más rápidos y con centros de rotación más estables (Figura 3B) en modelos experimentales tanto de FA,30 como de fibrilación ventricular (FV).29
Bloqueo de IK1 como estrategia antiarrítmica Dada la importante influencia de IK1 en la dinámica de rotores, su bloqueo resulta altamente atractivo para la terminación de la FA.31 En humanos el aumento de función de IK1 debido a una mutación en el gen KCNJ2, se asocia con FA familiar.32 A nivel experimental, el bloqueo de IK1 mediante cloruro de bario es capaz de detener la FV. Los actuales fármacos antiarrítmicos no tienen un papel preferencial sobre IK1, sin embargo, datos recientes publicados por Noujaim y colaboradores en cooperación con el grupo de Sánchez Chapula, han identificado la capacidad de la cloroquina para bloquear de forma preferencial IK1, lo cual se ha demostrado altamente eficaz en modelos animales de FA.31 Su capacidad antiarrítmica también se ha demostrado superior a la de la quinidina.33 Aunque en humanos su uso clínico no es como fármaco antiarrítmico, datos de los años 50's ya habían destacado su eficacia para restaurar el RS, en pacientes con FA crónica.34
A nivel teórico es posible el bloqueo de IK1 con selectividad auricular y bajo riesgo de afectación ventricular, basado en el perfil de expresión de la subfamilia Kir2.X de canales de K+ fuertemente–rectificadores de entrada. La expresión de la subfamilia Kir2.3 está mucho más concentrada en la aurícula humana que en los ventrículos.35
Corriente rectificadora de potasio dependiente de acetilcolina IK,ACh
De forma similar a la IK1, IK,ACh también ejerce importantes efectos en la dinámica de rotores. Su efecto a nivel celular se traduce en hiperpolarización del potencial de membrana diastólico y acortamiento en la duración del PA.19 Su efecto potenciador de la FA es ampliamente conocido por su uso en modelos experimentales clásicos de FA.9,20 Dicho efecto se traduce en un incremento en la frecuencia de rotación de los rotores (Figura 3C). El efecto producido es dosis dependiente, de tal forma que cuanto más alta sea la concentración de acetilcolina (ACh), más alta será la frecuencia de rotación del rotor y más complejo el patrón de conducción fibrilatoria.36 Esto último se debe a la aparición de heterogeneidad en la aurícula, debido a la distribución no homogénea de los receptores muscarínicos, a los cuales se une la ACh y de los canales iónicos Kir3.1 y Kir3.4 que se activan (Figura 2C, D y E). En estudios electrofisiológicos realizados en pacientes con FA paroxística, se observa un aumento en los valores de frecuencia dominante en aquellas zonas con valores basales más altos, en respuesta a la apertura de IK,ACh (IKAdo) tras la administración de adenosina.19 Recientemente se ha demostrado en muestras de cardiomiocitos humanos, la existencia de un incremento en la corriente IK,AChe IK1 en la aurícula izquierda de pacientes con FA paroxística, con presencia de un gradiente izquierda–derecha.37 Estos resultados podrían ayudar a explicar la presencia de rotores más estables en aurícula izquierda.
En pacientes con FA de larga evolución podría existir una disminución en el papel de IK,ACh, con una repuesta atenuada del PA a ACh en este contexto. No obstante, existen datos que sugieren que la forma constitutiva de la corriente IK,ACh (CA–IK,ACh,, activa en ausencia del agonista) (Figura 4), podría contribuir de forma importante al remodelado eléctrico relacionado con la FA.38 Los efectos en la prolongación del PA con el bloqueo de CA–IK,ACh podrían ser proporcionalmente mayores que en condiciones basales.
Bloqueo de la corriente IK,ACh
Debido a la que las subunidades alfa Kir3.X formadoras del poro se expresan predominantemente en las aurículas, el bloqueo de IK,ACh se convierte en una estrategia selectiva de bloqueo auricular.
Como soporte experimental el tertiapin–Q muestra un perfil de bloqueo bastante específico sobre las subunidades Kir3.X, con capacidad para terminar la FA en perros sometidos a estimulación auricular rápida. La terminación de la FA se asoció a prolongación del PA auricular y de la longitud de ciclo. La especificidad auricular se evidenció por la ausencia de la corriente a nivel de cardiomiocitos ventriculares, y por la ausencia de prolongación del PA ventricular.39 El tertiapin–Q es también capaz de bloquear CA–IK,ACh en el modelo canino. En células de aurícula derecha, en pacientes en FA, el tertiapin–Q es capaz de bloquear IK,ACh.40 Como contrapartida, el uso de tertiapin–Q podría asociarse a efectos adversos en el sistema nervioso central por sus acciones sobre corrientes iónicas y el PA neuronales. Por otro lado, al tratarse de una toxina procedente del veneno de abeja, existen dudas acerca de su aplicabilidad en humanos por su inmunogenicidad y unión a proteínas plasmáticas.41
Otras moléculas como NIP142 and NIP151 han demostrado varios grados de selectividad en el bloqueo de IK,ACh, con terminación de FA vagal en perros.42 Actualmente en estudio también se encuentra el NTC801, con un perfil de bloqueo selectivo de IK,ACh.
Fármacos de uso clínico y perfil de bloqueo multicanal también muestran bloqueo de IK,ACh. Destaca el efecto de la dronedarona con una potencia muy superior a la amiodarona sobre IK,ACh.43 También la flecainida podría llegar a bloquear la corriente IK,ACh, aunque la concentración inhibitoria del 50% de canales (IC50) fue 5.3 μM en células aisladas de cobaya,44 lo cual supone una concentración más de dos veces superior al límite terapéutico establecido en humanos.
Corriente rectificadora de potasio dependiente de ATP (IK,ATP)
En situaciones de isquemia, el descenso en el cociente ATP/APD abre el canal provocando un aumento en la conductancia de membrana al K+, descenso de la refractariedad y acortamiento en la duración del PA (Figura 3D).38 En preparaciones experimentales con muestras de corazones humanos, se observa un incremento en el riesgo de FA y FV al inducir un aumento de IK,ATP mediante la administración de pinacidil y diazóxido. Aunque el efecto de la apertura de IK,ATP parece incrementar el riesgo de FA, no está claro su papel en la FA paroxística ni en la persistente. Aunque con poca evidencia, podría existir una disminución de la corriente IKATP disponible en pacientes con FA permanente, lo cual sugeriría que la regulación de esta corriente no contribuye de forma relevante al remodelado iónico de la FA.45
Corriente de calcio tipo L (ICa–L)
La duración y la morfología del PA son altamente dependientes del flujo de Ca2+ a través de la membrana del cardiomiocito. ICa–L es necesaria para mantener la propagación del impulso, especialmente cuando dicha propagación es muy lenta, circunstancia que existe en el núcleo de los rotores, debido a la marcada curvatura del frente de onda y al potencial de membrana diastólico ligeramente despolarizado. El bloqueo de ICa–L mediante la administración de verapamilo aumenta el tamaño del núcleo del rotor, y disminuye la frecuencia de rotación y el grado de fragmentación de las ondas de propagación.46 En pacientes con FA persistente se ha descrito una reducción de los valores de la frecuencia dominante tras el tratamiento con verapamilo.47 Sin embargo, el potencial antiarrítmico de los agentes bloqueadores deICa–L se ve limitado por una disminución de su papel en FA persistente.48
Desde otra perspectiva, el Ca2+ también podría actuar como mecanismo de iniciación de la FA. Las alteraciones en la dinámica del Ca2+ intracelular se relacionan estrechamente con el desarrollo de pospotenciales tardíos, los cuales pueden dar origen a actividad desencadenada, así como al origen de un frente de onda que podría terminar en fragmentación e inicio de un rotor y FA. En modelos de aumento de presión intracavitaria se ha observado que la actividad focal, probablemente derivada de sobrecarga de Ca2+ y pospotenciales, es capaz de alterar la dinámica de los rotores, así como la generación de fragmentación de las ondas de propagación y formación de nuevos rotores.14
Corriente rectificadora tardía de potasio (IK)
La corriente IK tiene dos componentes: IKr e IKs, con rápida y lenta cinética de activación, respectivamente. En ambos casos, la cinética de desactivación muestra importantes variaciones entre especies. La combinación de lenta desactivación de IKr e IKs ayuda a explicar el mecanismo de refractariedad postrepolarización.49 La presencia de una distribución espacial heterogénea en la densidad de IK podría fácilmente explicar la formación de fragmentación del frente de onda, tanto a nivel auricular como ventricular. Durante una reentrada funcional, la reducción uniforme de IK debería incrementar la duración DPA de forma más marcada en la periferia, que en el centro de rotación. En las proximidades del núcleo, la repolarización ocurre de forma prematura debido a la influencia electrotónica del núcleo, lo cual devuelve el potencial de membrana al nivel de reposo antes de que IK tenga el tiempo suficiente para activarse. Por lo tanto, el bloqueo de IK no debería tener demasiado efecto sobre el núcleo del rotor, sin embargo, podría prolongar la DPA en la periferia del rotor dando lugar a conducción fibrilatoria.46
En monocapas con células ventriculares neonatales de rata, el aumento de IKr se asocia a rotores más rápidos, menor LO y PA más corto, lo que aumenta la estabilidad de los rotores.50 Estudios similares en monocapas de células que sobreexpresan IKs, han demostrado que una cinética de recuperación de IKs no homogénea en el espacio es suficiente para dar lugar a fragmentación de ondas y formación de reentrada.46
Corriente ultra–rápida rectificadora tardía de potasio (IKur)
Las subunidades Kv1.5 forman el canal proteico de la corriente IKur, que debido a su ausencia en los ventrículos (Figura 4) se convierte en diana para nuevos fármacos antiarrítmicos en FA.51 En cardiomiocitos humanos aislados, IKur muestra una lenta recuperación desde la inactivación a potenciales de membrana diastólicos y una dependencia de frecuencia significativa a temperaturas fisiológicas. El descenso de IKur con el aumento de frecuencia puede tender a mayor duración del PA en taquicardia, que si la corriente fuese independiente de la frecuencia. Con estas bases IKur podría ser protectora frente a arritmias por reentrada.
Bloqueo de la corriente IKur
Distintas sustancias han mostrado diferentes grados de selectividad en el bloqueo de IKur. El compuesto AVE0118 demostró bloqueo de IKur e ITo con concentraciones en rango μM similares, aunque también es capaz de bloquear IK,ACh, IKr e IKs con concentraciones más altas. Los resultados en modelo animal con remodelado eléctrico y estructural fueron inicialmente prometedores, observándose prolongación del periodo refractario auricular, terminación e incapacidad para la reinducción de FA, sin afectar la repolarización ventricular.52 No obstante, el desarrollo de la sustancia ha sido detenido en su fase clínica. Otras sustancias con analogía estructural han empezado a desarrollarse (AVE1231, AVE3295 y S9947).
Más dudas existen sobre si el bloqueo aislado de IKur es capaz de detener la FA. Datos experimentales y simulaciones apuntan a que en modelos de FA inducida con ACh o carbacol, el bloqueo selectivo de IKur no es capaz de restaurar el RS.30 El fracaso en la terminación, estudiado mediante simulaciones informáticas, se debe principalmente a la escasa contribución de IKur a la corriente neta de repolarización ante la presencia de alta contribución de IKACh.30 Añadido a esto, se ha observado un mayor riesgo de Fa en aurículas sanas de perro bajo bloqueo de IKur, lo cual tiene su correlación clínica con casos de FA familiar asociados a mutación en el gen KCNA5, que provoca una diminución de función del canal proteico de IKur.53
Corriente transitoria de salida de potasio(ITo)
En cardiomiocitos auriculares, la corriente ITo se activa en la fase de meseta del potencial de membrana y es responsable de la repolarización transitoria en la fase 1 del PA (Figura 4). La recuperación desde la inactivación ocurre de forma lenta, lo cual da lugar a baja amplitud durante frecuencias altas y prolongación del PA. En células ventriculares de conejo, ITo puede tener un papel en la aparición del fenómeno de Wenckebach.54 Esto último podría tener repercusiones en los bordes delimitando distintos dominios de frecuencia favoreciendo la conducción fibrilatoria, aunque otras corrientes como IK1 pueden tener un papel más dominante en la aparición de conducción fibrilatoria en los bordes. Su papel en la terminación de FA y dinámica de rotores no está completamente establecido, aunque el bloqueo combinado de ITo e IKur se ha demostrado superior al bloqueo de aislado de IK, en modelo animal de FA inducida mediante estimulación auricular.52
Bloqueo multicanal con selectividad auricular
La combinación de bloqueo de varias corrientes auriculares o perfiles de bloqueo multicanal con protección de efectos secundarios ventriculares, puede suponer una alternativa más evolucionada y eficaz para terminar la FA. Entre estas sustancias cabe destacar al vernakalant, recientemente aprobado para cardioversión farmacológica de FA paroxística, y a la ranolazina, actualmente aprobada para el control de la angina de pecho.
El vernakalant presenta un perfil de bloqueo de múltiples corrientes de predominio auricular: IKur, INa, IK,ACh, así como otras corrientes como IKr, ITo e ICa–L (Tabla 1).27,51,55 Este perfil multicanal podría explicar su alta eficacia en la cardioversión de FA paroxística. Sin embargo, el vernakalant no es superior a placebo en la conversión a RS de los pacientes con FA de más de siete días de duración.56 En FA de larga duración, el aumento de la influencia de las corrientes rectificadoras de K+ como IK1 y CA–IK,ACh, podría influir en la pérdida de efecto antiarrítmico en esta población de pacientes. Su administración se restringe a la vía intravenosa, lo cual supone otra limitación para evaluar su eficacia en la prevención de recurrencias. Aunque una nueva formulación apta para administración oral está en proceso de evaluación, tendremos que esperar a que aparezcan los resultados clínicos para establecer su utilidad.
La ranolazina ha demostrado ser efectiva en la terminación y prevención de FA, en distintos modelos experimentales. A concentraciones de 10 μM es capaz de terminar y prevenir la FA vago–dependiente, o la FA con estimulación β–adrenérgica en modelo de isquemia y reperfusión. También es capaz de bloquear la generación de pospotenciales, tanto precoces como tardíos producidos por incremento en la corriente tardía de Na+.26
El mecanismo por el que la ranolazina ejerce sus efectos antiarrítmicos incluye distintos aspectos. Por un lado disminuye el pico de INa, reduciendo la excitabilidad y provocando un aumento del periodo refractario efectivo con imposibilidad para activación de la aurícula a altas frecuencias.57 El efecto sobre I ,58–61 combinado con el bloqueo de IKr58,62 tiene una acción sinérgica al reducir el intervalo diastólico y potenciar el efecto sobre el bloqueo de INa, al tratarse del momento en el que el canal de Na+ se recuperaría del bloqueo (Tabla 1). El efecto combinado se traduce en una menor cantidad de Ca2+ intracelular, suprimiendo así la aparición de pospotenciales. El papel sobre la corriente tardía de Na+ en el contexto de la FA es menos evidente. La ranolazina podría contribuir a la supresión de pospotenciales que actuarían como desencadenantes, al reducir el Ca2+ intracelular de forma secundaria al descenso de la actividad intracelular del Na+. Esto supone un mecanismo protector para la aparición de arritmias ventriculares, en el contexto de bloqueo de IKr. El bloqueo de la corriente tardía de Na+ tiene especial importancia en el contexto de patologías con prolongación de la repolarización en los ventrículos, como el síndrome de QT largo61 o la insuficiencia cardiaca. En ambos casos, la normalización de la repolarización prevendría la aparición de arritmias.26 Los datos clínicos disponibles hasta el momento sugieren que la ranolazina puede ser segura y efectiva en la supresión de FA, y probablemente en ciertos tipos de arritmias ventriculares, aunque son necesarios más datos procedentes de ensayos a gran escala y aleatorizados para completar su perfil clínico y de seguridad.
Bloqueo multicanal en base a fármacos de eficacia demostrada
Este grupo de sustancias se basan fundamentalmente en la eficacia antiarrítmica de la amiodarona para terminar la FA, lo cual se asocia a un perfil de seguridad muy alto en la mayoría de cardiopatías. Como contrapartida se encuentran los efectos adversos extracardíacos derivados de su uso a medio–largo plazo,63 motivo por el que se buscan nuevas sustancias análogas a la amiodarona.
La dronedarona, actualmente aprobada para su uso a nivel clínico, es un derivado no iodado de la amiodarona con perfil de bloqueo multicanal y acción antiadrenérgica no competitiva. Aunque su perfil electrofisiológico in vitro es similar al de la amiodarona, la dronaderona es hasta 10 veces más potente en el bloqueo de INa en células auriculares humanas, hasta 100 veces más potente en el bloqueo de IK,ACh en células auriculares de cobaya y tiene un mayor efecto antiadrenérgico (Tabla 2).43

La dronedarona ha demostrado ser significativamente más efectiva que placebo en el mantenimiento del RS en pacientes con FA, sumado a escasa toxicidad extracardíaca. Al mismo tiempo tiene capacidad para el control de frecuencia durante FA. No obstante, su eficacia para detener la FA a nivel experimental parece ser inferior a la de la amiodarona,64 así como su capacidad para reducir recurrencias a nivel clínico.65 La superioridad de la amiodarona podría ser debida a múltiples aspectos entre los cuales está un diferente perfil en el bloqueo multicanal (Tabla 2), aunque podríamos destacar la inducción de refractariedad postrepolarización con amiodarona, que no se ha demostrado bajo dronedarona. El uso de dronedarona se ha asociado a la aparición de nuevos efectos adversos con necesidad de especial precaución en pacientes con insuficiencia cardiaca en clase funcional III–IV de la NYHA, en los que se ha demostrado un aumento de la mortalidad en el ensayo clínico ANDROMEDA.66 Entre las posibles razones para este incremento en mortalidad podría encontrarse su potente acción bloqueadora de INa (Tabla 2), asociada al bloqueo de ICa–L lo cual podría deteriorar el cuadro de insuficiencia cardiaca. Recientemente también han sido descritos casos aislados de daño hepático severo,67 recomendándose la monitorización de enzimas hepáticas. Hoy en día, la utilidad clínica y perfil de seguridad de la dronedarona no está definido por completo, lo cual ha llevado a la FDA (Food and Drug Administration) a restringir su uso a la prevención de recurrencias en pacientes con FA no permanente, y sin cardiopatía estructural.
La budiodarona es un análogo químico de la amiodarona con perfil de bloqueo de canales de Na+, K+ y Ca2+ similar a la amiodarona. Datos iniciales en pacientes con FA paroxística han demostrado una reducción de la carga de FA, mediante una reducción en la duración media de los episodios.68 Más datos procedentes de ensayos clínicos a mayor escala son necesarios para sacar conclusiones sobre su papel en la FA.
La celivarona es un componente no iodado estructuralmente relacionado con amiodarona y dronedarona. Es capaz de bloquear las corrientes IKr, IKs, IK,ACh IKur, ICa–L, y disminuye la amplitud de Ca2+ intracelular y la contracción celular. Muestra bloqueo de la respuesta adrenérgica tanto α–1 como β–1, así como del receptor AT1 de angiotensina. En modelos animales ha demostrado capacidad para terminar la FA y prevenir la reinducción. Su evaluación en humanos se encuentra en la fase inicial.69
El compuesto AVE1231 representa otro ejemplo de sustancia con bloqueo multicanal derivada del AVE0118, el cual aunque sí demostró eficacia para terminar la FA en varios estudios experimentales, sus resultados clínicos no fueron satisfactorios. Esto se unió a un espectro limitado en el tratamiento a largo plazo de la FA, debido al filtro hepático de primer paso. Nuevos datos serán necesarios para aclarar la eficacia de AVE1231 y su mejoría en el perfil clínico.
Otras estrategias antiarrítmicas novedosas
Regulación de la homeostasis del calcio intracelular La alteración en la regulación del Ca2+ intracelular se relaciona con el desarrollo de FA. De forma opuesta, la normalización de la liberación de Ca2+ del retículo sarcoplásmico (RSarc) se perfila como una estrategia terapéutica teórica. Distintos componentes están implicados en la regulación del Ca2+ intracelular: el RSarc, el canal de liberación de Ca2+, también conocido como receptor de rianodina (RyR2), la proteína accesoria calstabina 2 (subu–nidad inhibidora del receptor RyR2; FKBP12.6) de unión a RyR2 y la ATPasa de Ca2+ del RSarc (SERCA2A). La alteración en la regulación del Ca2+ intracelular puede promover FA, mediante su implicación en la generación de pospotenciales tanto tardíos como precoces, y los conocidos como potenciales precoces al final de la fase 3 del PA, que pueden dar lugar a actividad desencadenada. Aunque la aparición de pospotenciales juega un papel claro como desencadenante de FA, más discutible es su importancia en el mantenimiento a largo plazo de la arritmia. Otros aspectos atribuibles a la alteración de la regulación de Ca2+ intracelular, se relacionan con la modulación del remodelado eléctrico y su implicación en numerosas cascadas enzimáticas involucradas en el remodelado estructural (Figura 5).70
El defecto en la regulación del receptor RyR2 origina un incremento de la liberación de Ca2+ diastólico, que se relaciona con el desarrollo de FA. Este mecanismo ha sido descrito en cardiomiocitos aislados de pacientes con FA de larga duración (>6 meses), así como en perros con remodelado auricular inducido por estimulación auricular a alta frecuencia. La liberación de Ca2+ diastólico podría ser debida a hiperfosforilación del receptor RyR2 mediada por la proteína quinasa–A, lo cual provoca una disociación de la subunidad estabilizadora calstabina 2 y un aumento en la probabilidad del estado abierto del receptor RyR2.71 En modelo de ratón se ha demostrado que la deficiencia de calstabina 2 favorece la inducción de FA, asociado a mayor liberación de Ca2+ desde el RSarc, comparado con el grupo control.72
Desde el punto de vista terapéutico resulta complejo regular la homeostasis del Ca2+ intracelular, sin comprometer la contractilidad miocárdica. Sustancias como la JTV519 (o K201) ha demostrado capacidad para disminuir la inducibilidad de FA en perros. Esto se atribuye a un mecanismo de facilitación de la unión de calstabina 2 al receptor RyR2 fosforilado vía la proteína quinasa–A, asociado a un perfil de bloqueo multicanal de INa, ICa–L, IKr, IK1 e IK,ACh con prolongación del periodo refractario efectivo.73 ' Nuevas sustancias y derivados necesitan ser desarrollados para aumentar su especificidad e investigar su perfil clínico.
Terapia de regulación de las uniones abiertas
Las uniones abiertas son canales que conectan directamente el interior de células cardíacas adyacentes, y permiten la propagación intercelular de corriente eléctrica a través de vías de baja resistencia. Están formadas por proteínas llamadas conexinas (Cx), que son complejos hexaméricos que normalmente se congregan en los discos intercalares de las células miocárdicas. La Cx43, Cx45 y Cx40 se expresan en el corazón, con predominio de Cx40 en el tejido auricular. La Cx40 no se encuentra en el miocardio ventricular, pero sí en el tejido de conducción ventricular.74 Su importancia en la generación de arritmias radica en que la afectación del acoplamiento celular puede disminuir la VC y causar un sustrato heterogéneo para la generación de reentrada (Figura 5). Durante isquemia, el desacoplamiento celular por defosforilación y cierre parcial de uniones abiertas se considera un mecanismo importante en la aparición de retraso en la conducción. En esta línea, el rotigaptide tiene perfil antiarrítmico en modelos de FA en contexto de isquemia aguda, posiblemente en parte mediante mejoría de la conducción a través de las uniones abiertas.75 El GAP–134, con un perfil similar al rotigaptide, también ha demostrado mejorar la VC y reducir la carga de FA en modelos caninos de FA.76
La relevancia clínica de una estrategia basada en la mejoría de la conducción a través de las uniones abiertas todavía tiene que ser evaluada.
Terapias de control del sustrato arritmogénico auricular
El remodelado auricular constituye una serie de cambios tanto a nivel estructural como iónico y mecánico, que favorecen el principio y perpetuación de la FA.7,8 Su presencia explica las dificultades para restaurar el RS en pacientes con FA de larga duración, así como la alta tasa de recurrencia tras la cardioversión. A lo largo de semanas se observa una disminución de ICa–L, ITo e IKur, junto con aumento de las corrientes rectificadoras de K+, IK1 y CA–IKACh (Figura 6).48 Estos cambios acortan la duración del PA y el periodo refractario efectivo auricular.77 Además, se favorece la presencia de un sustrato heterogéneo al ocurrir con una magnitud diferente en distintas zonas de las aurículas (Figura 5). Alteraciones en la homeostasis del Ca2+ provocan alteraciones en las propiedades contráctiles del tejido auricular y dilatación. De forma paralela, la disminución en la expresión de Cx, junto con el desarrollo progresivo de fibrosis afecta a las propiedades de conducción, que se convierte en más lenta y fraccionada.78 El conjunto de cambios favorece tanto la generación de rotores como su persistencia.
Enfermedades diversas como la hipertensión arterial, la insuficiencia cardiaca o cardiopatía isquémica, entre otras, pueden inducir los cambios descritos. En este contexto, las terapias para prevención o retraso del remodelado auricular tienen como objetivo controlar la carga de FA (Figura 5). La mayor parte de la evidencia científica se centra en los fármacos inhibidores del sistema renina–angiotensina–aldosterona (SRA), como los inhibidores de la enzima conversora de la angiotensina (IECAs) o los bloqueadores del receptor AT2 de la angiotensina (ARA–II). Menos evidencia existe en favor de terapias como los ácidos grasos poliinsaturados omega 3, estatinas y glucocorticoides. Resultados de estudios en experimentación animal y pacientes con cardesantán e irbesartan, respectivamente, sugieren que su uso previene el desarrollo de fibrosis y FA en el modelo animal, así como la recurrencia precoz tras cardioversión en pacientes con FA.79 Datos procedentes de un modelo experimental en perros con estimulación auricular a alta frecuencia y ablación del nodo AV para control de la frecuencia ventricular, muestran datos opuestos, sin efecto del enalapril en la prevención de fibrosis y sin mayor fibrosis en el grupo control.80 El estudio GISI–AF con 1142 pacientes pacientes aleatorizados a valsartan o placebo, con el objetivo de valorar la tasa de recurrencia de FA tras cardioversión, durante un periodo de seguimiento a un año, muestra que valsartán no reduce la recurrencia de episodios de FA en pacientes preferentemente hipertensos y con función sistólica del ventrículo izquierdo >40%.81
La confusión en los resultados de estos estudios podría ser debida a que en presencia de factores que promueven la ibrosis auricular, como la insuiciencia cardiaca o taquimiocardiopatía por frecuencias ventriculares rápidas, la prevención de fibrosis podría tener un factor protector en el remodelado estructural. Por el contrario, ante la ausencia de factores que promueven la fibrosis, como en el modelo animal con ablación de nodo AV para control de la frecuencia ventricular o en los pacientes con buena función ventricular del estudio GISSI–AF, el escaso desarrollo de fibrosis no se mejora con añadir terapias preventivas. Esta visión se refuerza con datos procedentes de modelo animal ovino con y sin ablación del nodo AV, para control de la frecuencia ventricular. Las ovejas con estimulación auricular rápida y sin ablación de nodo AV, desarrollan mayor fibrosis y FA de forma más precoz. El uso de inhibidores del SRA (quinaprilo y losartán) previene o retrasa el desarrollo de FA persistente, mediante el control de la fibrosis auricular.82 Algunos ensayos clínicos han mostrado que los ARA–II y las estatinas podrían tener especial beneficio en los pacientes con FA y disfunción sistólica de ventrículo izquierdo, sin beneficio en aquellos pacientes sin cardiopatía.83 Recientemente, los datos de un estudio retrospectivo en 83 pacientes también sugieren que la espironolactona es capaz de disminuir de forma significativa la carga de FA, en pacientes con cardiopatía estructural, comparado con el grupo bajo terapia convencional.84
El desarrollo de fibrosis a nivel auricular es un proceso multifactorial en el que intervienen diversos procesos neurohormonales y mediadores celulares. Como ejemplo, la hipertrofia en cardiomiocitos y la fibrosis se correlacionan fuertemente con un incremento en la expresión de TGFb1. El bloqueo de este mediador o algún punto de su cascada metabólica podría ser una vía para la regulación de la fibrosis auricular.85
Múltiples ensayos clínicos en curso están tratando de controlar y entender el papel de la fibrosis y sus mediadores en el desarrollo de FA. La estrategia se presenta especialmente atractiva al suponer una aproximación real al control del sustrato estructural, que probablemente perpetúe la presencia de FA en muchos pacientes con FA de larga duración.
Direcciones futuras en fármacos antiarrítmicos
El objetivo final de los fármacos antiarrítmicos en FA sería el control absoluto de la arritmia. Esto podría actuar sobre la principal causa de aumento de mortalidad en esta población, como son los eventos cardioembólicos.86
El nuevo desarrollo de fármacos con selectividad auricular es una vía que se prevé altamente atractiva en los próximos años, sobre todo en la vertiente de bloqueo multicanal con especificidad auricular. Poblaciones concretas de pacientes como aquellos con FA familiar o determinadas circunstancias isiopatológicas, en las que se identifique un mecanismo iónico preferencial podrán beneficiarse del bloqueo selectivo de esa corriente iónica. Del mismo modo, se ha demostrado que el tipo de cardiopatía de base influye en el tipo de remodelado iónico, sin un patrón fijo de remodelado que haya prever que un mismo fármaco pueda ser eicaz para todos los casos de FA. El conocimiento profundo de los mecanismos del proceso de remodelado de determinadas cardiopatías permitirá usar los fármacos disponibles de una forma más eicaz, y al mismo tiempo desarrollar nuevas sustancias más específicas.
Especialmente relevantes se muestran las terapias para prevención o retraso del remodelado auricular. Limitaciones al uso de fármacos antiarrítmicos como el tope de dosis, sin obtener los resultados esperados podrían ser adecuadas para actuar sobre el sustrato auricular y así potenciar el efecto antiarrítmico del fármaco seleccionado. La selección de candidatos para terapias reguladoras del sustrato debe centrarse en los pacientes en que el sustrato es relevante, y por lo tanto requieren identiicar las poblaciones de riesgo.
Conclusiones
El estudio y profundización en el entendimiento de los mecanismos básicos que dan comienzo y mantienen la FA, tanto desde el punto de la dinámica de propagación de ondas y patrones de activación, como iónico y estructural, ha supuesto el relanzamiento de nuevas estrategias antiarrítmicas, focalizadas en detener o mitigar desde distintos frentes, los principales factores implicados en su perpetuación. Se ha conseguido una mayor selectividad auricular en el efecto antiarrítmico, aunque los retos derivados de nuevos efectos secundarios y la complejidad clínica de la arritmia hacen que la FA se mantenga como uno de los principales retos para múltiples grupos de investigación a nivel internacional.
Agradecimientos
Este trabajo se condujo en parte bajo de los auspicios de subvenciones del USA National Institutes of Health (P01–HL039707 and P01–HL087226), de la Fundación Leducq (J.J) y la Fundación Alfonso Martín Escudero (DFR).
Referencias
1. Kannel WB, Wolf PA, Benjamin EJ, et al. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population–based estimates. Am J Cardiol 1998;82:2N–9N. [ Links ]
2. Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial fibrillation (ATRIA) Study. JAMA 2001;285:2370–2375. [ Links ]
3. Merino JL, Doiny D, Estrada A, et al. Repercusión de la fibrilación auricular: Epidemiología e impacto clínico–económico. RIA 2011;1:246–258. [ Links ]
4. Marquez MF, Gomez–Flores J, Aranda–Faustro A, et al. Recientes avances en la fisiopatología de la fibrilación auricular. Arch Cardiol Mex 2009;79 Suppl 2:18–25. [ Links ]
5. Lafuente–Lafuente C, Mouly S, Longas–Tejero MA, et al. Antiarr–hythmic drugs for maintaining sinus rhythm after cardioversion of atrial fibrillation: a systematic rev'ew of randomized controlled trials. Arch Intern Med 2006;166:719–28. [ Links ]
6. Kober L, Torp–Pedersen C, McMurray JJ, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008;358:2678–2687. [ Links ]
7. Wijffels MC, Kirchhof CJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954–1968. [ Links ]
8. Morillo CA, Klein GJ, Jones DL, et al. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995;91:1588–1595. [ Links ]
9. Moe GK, Abildskov JA. Atrial fibrillation as a self–sustaining arrhythmia independent of focal discharge. Am Heart J 1959;58:59–70. [ Links ]
10. Moe GK, Rheinboldt WC, Abildskov JA. A Computer Model of Atrial fibrillation. Am Heart J 1964;67:200–220. [ Links ]
11. Jalife J. ¿Por qué es la Aurícula Izquierda tan Importante en el Mecanismo de la Fibrilación Auricular Crónica? RIA 2009;1:3–26. [ Links ]
12. Kalifa J, Jalife J, Zaitsev AV, et al. Intra–atrial pressure increases rate and organization of waves emanating from the superior pulmonary veins during atrial fibrillation. Circulation 2003;108:668–671. [ Links ]
13. David Filgueiras Rama, José Jalife. Mechanisms Underlying Atrial fibrillation. In: Charles Antzelevitch, ed. Basic Science for Clinical Electrophysiologist. Philadelphia, USA. Saunders. 2011. 141156. [ Links ]
14. Yamazaki M, Vaquero LM, Hou L, et al. Mechanisms of stretch–induced atrial fibrillation in the presence and the absence of adrenocholinergic stimulation: interplay between rotors and focal discharges. Heart Rhythm 2009;6:1009–1017. [ Links ]
15. Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of trachycardia. Circ Res 1973;33:54–62. [ Links ]
16. Klos M, Calvo D, Yamazaki M, et al. Atrial septopulmonary bundle of the posterior left atrium provides a substrate for atrial fibrillation initiation in a model of vagally mediated pulmonary vein tachycardia of the structurally normal heart. CircArrhythmElectrophysiol 2008;1:175–183. [ Links ]
17. Cabo C, Pertsov AM, Davidenko JM, et al. Vortex shedding as a precursor of turbulent electrical activity in cardiac muscle. Biophys J 1996;70:1105–1111. [ Links ]
18. Wellner M, Berenfeld O, Jalife J, et al. Minimal principle for rotor filaments. Proc Natl Acad Sci U S A 2002;99:8015–8018. [ Links ]
19. Atienza F, Almendral J, Moreno J, et al. Activation of inward rectiier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation 2006;114:2434–2442. [ Links ]
20. Berenfeld O, Mandapati R, Dixit S, et al. Spatially distributed dominant excitation frequencies reveal hidden organization in atrial fibrillation in the Langendorff–perfused sheep heart. J Cardiovasc Electrophysiol 2000;11:869–879. [ Links ]
21. Kalifa J, Tanaka K, Zaitsev AV, et al. Mechanisms of wave fractionation at boundaries of high–frequency excitation in the posterior left atrium of the isolated sheep heart during atrial fibrillation. Circulation 2006;113:626–633. [ Links ]
22. Jalife J, Pandit SV. Ionic mechanisms of wavebreak in fibrillation. Heart Rhythm 2005;2:660–663. [ Links ]
23. Kneller J, Kalifa J, Zou R, et al. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically–realistic mathematical model. Circ Res 2005;96:e35–47. [ Links ]
24. Donovan KD, Power BM, Hockings BE, et al. Intravenous flecainide versus amiodarone for recent–onset atrial fibrillation. Am J Cardiol 1995;75:693–697. [ Links ]
25. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med 1989;321:406–412. [ Links ]
26. Antzelevitch C, Burashnikov A, Sicouri S, et al. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heartrhythm 2011;8:1281–1290. [ Links ]
27. Fedida D, Orth PM, Chen JY, et al. The mechanism of atrial antiarrhythmic action of RSD1235. J CardiovascElectrophysiol 2005;16:1227–1238. [ Links ]
28. Samie FH, Berenfeld O, Anumonwo J, et al. Rectiication of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res 2001;89:1216–1223. [ Links ]
29. Noujaim SF, Pandit SV, Berenfeld O, et al. Up–regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol 2007;578:315–326. [ Links ]
30. Pandit SV, Zlochiver S, Filgueiras–Rama D, et al. Targeting atrioventricular differences in ion channel properties for terminating acute atrial fibrillation in pigs. Cardiovasc Res 2011;89:843–851. [ Links ]
31. Noujaim SF, Stuckey JA, Ponce–Balbuena D, et al. Speciic residues of the cytoplasmic domains of cardiac inward rectiier potassium channels are effective antiibrillatory targets. FASEB J 2010;24:4302–4312. [ Links ]
32. Xia M, Jin Q, Bendahhou S, et al. A Kir2.1 gain–of–function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun 2005;332:1012–1019. [ Links ]
33. Noujaim SF, Stuckey JA, Ponce–Balbuena D, et al. Structural bases for the different anti–ibrillatory effects of chloroquine and quinidine. Cardiovasc Res 2011;89:862–869. [ Links ]
34. Burrell ZL, Jr., Martinez AC. Chloroquine and hydroxychloroquine in the treatment of cardiac arrhythmias. N Engl J Med 1958;258:798–800. [ Links ]
35. Wang Z, Yue L, White M, et al. Differential distribution of inward rectiier potassium channel transcripts in human atrium versus ventricle. Circulation 1998;98:2422–2428. [ Links ]
36. Berenfeld O. Ionic and substrate mechanism of atrial fibrillation: rotors and the exitacion frequency approach. Arch Cardiol Mex 2010;80:301–314. [ Links ]
37. Voigt N, Trausch A, Knaut M, et al. Left–to–right atrial inward rectiier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. CircArrhythmElectrophysiol 2010;3:472–480. [ Links ]
38. Ehrlich JR. Inward rectiier potassium currents as a target for atrial fibrillation therapy. J Cardiovasc Pharmacol 2008;52:129–135. [ Links ]
39. Cha TJ, Ehrlich JR, Chartier D, et al. Kir3–based inward rectiier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation 2006;113:1730–1737. [ Links ]
40. Dobrev D, Friedrich A, Voigt N, et al. The G protein–gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 2005;112:3697–3706. [ Links ]
41. Jin W, Lu Z. A novel high–afinity inhibitor for inward–rectiier K+ channels. Biochemistry 1998;37:13291–13299. [ Links ]
42. Nattel S, Carlsson L. Innovative approaches to anti–arrhythmic drug therapy. Nat Rev Drug Discov 2006;5:1034–1049. [ Links ]
43. Patel C, Yan GX, Kowey PR. Dronedarone. Circulation 2009;120:636–644. [ Links ]
44. Inomata N, Ishihara T, Akaike N. Mechanisms of the anticholinergic effect of SUN 1165 in comparison rnth flecainide, disopyramide and quinidine in single atrial myocytes isolated from guinea–pig. Br J Pharmacol 1991;104:1007–1011. [ Links ]
45. Balana B, Dobrev D, Wettwer E, et al. Decreased ATP–sensitive K(+) current density during chronic human atrial fibrillation. JMCC 2003;35:1399–1405. [ Links ]
46. Samie FH, Mandapati R, Gray RA, et al. A mechanism of transition from ventricular fibrillation to tachycardia : effect of calcium channel blockade on the dynamics of rotating waves. Circ Res 2000;86:684–691. [ Links ]
47. Bollmann A, Sonne K, Esperer HD, et al. Patients with persistent atrial fibrillation taking oral verapamil exhibit a lower atrial frequency on the ECG. Ann Noninvasive Electrocardiol 2002;7:92–97. [ Links ]
48. Pandit SV, Berenfeld O, Anumonwo JM, et al. Ionic determinants of functional reentry in a 2–D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J2005;88:3806–3821. [ Links ]
49. Munoz V, Grzeda KR, Desplantez T, et al. Adenoviral expression of IKs contributes to wavebreak and ibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res 2007;101:475–483. [ Links ]
50. Hou L, Deo M, Furspan P, et al. A major role for HERG in determining frequency of reentry in neonatal rat ventricular myocyte monolayer. Circ Res 2010;107:1503–1511. [ Links ]
51. Ehrlich JR, Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs 2009;69:757–774. [ Links ]
52. Blaauw Y, Gogelein H, Tieleman RG, et al. "Early" class III drugs for the treatment of atrial fibrillation: eficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation 2004;110:1717–1724. [ Links ]
53. Olson TM, Alekseev AE, Liu XK, et al. Kv1.5 channelopathy due to KCNA5 loss–of–function mutation causes human atrial fibrillation. Hum Mol Genet 2006;15:2185–2191. [ Links ]
54. Yehia AR, Shrier A, Lo KC, et al. Transient outward current contributes to Wenckebach–like rhythms in isolated rabbit ventricular cells. Am J Physiol 1997;273:H1–11. [ Links ]
55. Fedida D. Vernakalant (RSD1235): a novel, atrial–selective antii–brillatory agent. ExpertOpinInvestigDrugs 2007;16:519–532. [ Links ]
56. Stiell IG, Roos JS, Kavanagh KM, et al. A multicenter, open–label study of vernakalant for the conversion of atrial fibrillation to sinus rhythm. Am Heart J 2010;159:1095–1101. [ Links ]
57. Sossalla S, Kallmeyer B, Wagner S, et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol 2010;55:2330–2342. [ Links ]
58. Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electro–physiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 2004;110:904–910. [ Links ]
59. Undrovinas AI, Belardinelli L, Undrovinas NA, et al. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J CardiovascElectrophysiol 2006;17 Suppl 1:S169–S77. [ Links ]
60. Burashnikov A, Di Diego JM, Zygmunt AC, et al. Atrium–selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 2007;116:1449–1457. [ Links ]
61. Fredj S, Sampson KJ, Liu H, et al. Molecular basis of ranolazine block of LQT–3 mutant sodium channels: evidence for site of action. Br J Pharmacol 2006;148:16–24. [ Links ]
62. Rajamani S, Shryock JC, Belardinelli L. Rapid kinetic interactions of ranolazine with HERG K+ current. J CardiovascPharmacol 2008;51:581–589. [ Links ]
63. Colin Lizalde LJ, Febles Palenzuela T, Crespo Mancebo F, et al. Un estudio retrospectivo para el análisis de los efectos colaterales de la amiodarona. Inst Cardiol Mex 1990;60:455–460. [ Links ]
64. Burashnikov A, Belardinelli L, Antzelevitch C. Acute dronedarone is inferior to amiodarone in terminating and preventing atrial fibrillation in canine atria. Heartrhythm 2010;7:1273–1279. [ Links ]
65. Le Heuzey JY, De Ferrari GM, Radzik D, et al. A short–term, randomized, double–blind, parallel–group study to evaluate the eficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J CardiovascElectrophysiol 2010;21:597–605. [ Links ]
66. Kober L, Torp–Pedersen C, McMurray JJ, et al. Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med 2008;358:2678–2687. [ Links ]
67. Joghetaei N, Weirich G, Huber W, et al. Acute liver failure associated with dronedarone. CircArrhythmElectrophysiol 2011;4:592–593. [ Links ]
68. Arya A, Silberbauer J, Teichman SL, et al. A preliminary assessment of the effects of ATI–2042 in subjects with paroxysmal atrial fibrillation using implanted pacemaker methodology. Europace 2009;11:458–464. [ Links ]
69. Gautier P, Serre M, Cosnier–Pucheu S, et al. In vivo and in vitro antiarrhythmic effects of SSR149744C in animal models of atrial fibrillation and ventricular arrhythmias. J CardiovascPharmacol 2005;45:125–135. [ Links ]
70. Dobrev D, Nattel S. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J CardiovascPharmacol 2008;52:293–299. [ Links ]
71. Vest JA, Wehrens XH, Reiken SR, et al. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005;111:2025–2032. [ Links ]
72. Sood S, Chelu MG, van Oort RJ, et al. Intracellular calcium leak due to FKBP12.6 deiciency in mice facilitates the inducibility of atrial fibrillation. Heartrhythm 2008;5:1047–1054. [ Links ]
73. Kumagai K, Nakashima H, Gondo N, et al. Antiarrhythmic effects of JTV–519, a novel cardioprotective drug, on atrial fibrillation/ flutter in a canine sterile pericarditis model. J CardiovascElectrophysiol 2003;14:880–884. [ Links ]
74. Chaldoupi SM, Loh P, Hauer RN, et al. The role of connexin40 in atrial fibrillation. Cardiovasc Res 2009;84:15–23. [ Links ]
75. Shiroshita–Takeshita A, Sakabe M, Haugan K, et al. Model–dependent effects of the gap junction conduction–enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 2007;115:310–318. [ Links ]
76. Laurent G, Leong–Poi H, Mangat I, et al. Effects of chronic gap junction conduction–enhancing antiarrhythmic peptide GAP–134 administration on experimental atrial fibrillation in dogs. CircA–rrhythmElectrophysiol 2009;2:171–178. [ Links ]
77. Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. CircArrhythmElectrophy–siol 2008;1:62–73. [ Links ]
78. Zlochiver S, Munoz V, Vikstrom KL, et al. Electrotonic myoi–broblast–to–myocyte coupling increases propensity to reentrant arrhythmias in two–dimensional cardiac monolayers. Biophys J 2008;95:4469–4480. [ Links ]
79. Kumagai K, Nakashima H, Urata H, et al. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am CollCardiol 2003;41:2197–2204. [ Links ]
80. Shinagawa K, Mitamura H, Ogawa S, et al. Effects of inhibiting Na(+)/H(+)–exchange or angiotensin converting enzyme on atrial tachycardia–induced remodeling. Cardiovasc Res 2002;54:438–446. [ Links ]
81. Disertori M, Latini R, Barlera S, et al. Valsartan for prevention of recurrent atrial fibrillation. N Engl J Med 2009;360:1606–1617. [ Links ]
82. Anne W, Willems R, Holemans P, et al. Self–terminating AF depends on electrical remodeling while persistent AF depends on additional structural changes in a rapid atrially paced sheep model. J Mol CellCardiol 2007;43:148–158. [ Links ]
83. Healey JS, Baranchuk A, Crystal E, et al. Prevention of atrial fibrillation with angiotensin–converting enzyme inhibitors and angiotensin receptor blockers: a meta–analysis. J Am CollCardiol 2005;45:1832–1839. [ Links ]
84. Williams RS, deLemos JA, Dimas V, et al. Effect of spironolactone on patients with atrial fibrillation and structural heart disease. Clin Cardiol 2011;34:415–419. [ Links ]
85. Sakata Y, Chancey AL, Divakaran VG, et al. Transforming growth factor–beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac–restricted overexpression of tumor necrosis factor. Basic Res Cardiol 2008;103:60–68. [ Links ]
86. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991;22:983–988. [ Links ]

