











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los grandes cambios (económicos, sociales, políticos y culturales) que han ocurrido en México durante los últimos 50 años han modificado las tendencias nutricionales y epidemiológicas. La adopción de un estilo de vida de occidental, incluyendo cambios drásticos en la dieta y en la realización de actividad física han conllevado el incremento en la prevalencia de factores de riesgo cardiometabólicos (dislipidemia, hipertensión, tabaquismo, diabetes, obesidad y sedentarismo), favoreciendo así un incremento rápido en las enfermedades crónicas asociadas a la obesidad1.
El tejido adiposo es el principal sitio de almacenamiento de triglicéridos en el organismo y está compuesto por adipocitos maduros y los diferentes tipos celulares que comprenden a la fracción del estroma vascular2. El cuerpo humano tiene diversos tipos de tejido adiposo, estos incluyen el tejido adiposo pardo, y el tejido adiposo blanco, que se divide en tejido adiposo subcutáneo y en tejido adiposo visceral (TAV), el cual está adosado a los órganos. El tejido adiposo blanco es considerado un órgano endócrino altamente activo y dentro de sus principales productos de secreción se encuentran las adipocinas3 4.
El TAV secreta adipocinas como la leptina, quemerina, resistina, visfatina, adiponectina, angiotensinógeno, apelina y omentina, además de citocinas inflamatorias; también está involucrado en la secreción de moléculas protrombóticas y quimiocinas proinflamatorias, como el inhibidor del activador del plasminógeno-1 y proteína quimiotáctica de monocitos 1(MCP-1), respectivamente5 6.
Estas adipocinas pueden modular la función de la vasculatura, también regulan el metabolismo de los hidratos de carbono y de los lípidos, y desempeñan un papel importante en la patogénesis de la resistencia a la insulina, así como también en el proceso inflamatorio. Como órgano dinámico, los cambios dentro de la función o anatomía celular de los adipocitos son estímulos para la modificación del perfil de adipocinas6 7. El objetivo de esta revisión es describir la participación de la omentina en las complicaciones metabólicas y cardiovasculares relacionadas con la obesidad.
Omentina
La omentina fue previamente descrita como intelectina-1 y fue encontrada en el intestino delgado en células de Paneth de ratón, células endoteliales, células epiteliales, enterocitos del intestino delgado y células estromales vasculares. También ha sido identificada en el pulmón, corazón, colon y timo, y se expresa de manera significativa en el TAV epicárdico y de las arterias coronarias8 9. Sus acciones están relacionadas con la estimulación de la sensibilización a la insulina en los adipocitos viscerales. Por otro lado, también puede potenciar la acción de la insulina en el tejido adiposo subcutáneo y está asociada a la modulación de la respuesta inflamatoria innata8 9.
La omentina es sintetizada y secretada, principalmente, en las células de la fracción estromal vascular del TAV (compuesto por diferentes tipos celulares tales como preadipocitos, células madre mesenquimales, células progenitoras endoteliales, macrófagos, linfocitos B y linfocitos T). Como factor de secreción, la omentina puede ser una hormona que ejerce un mecanismo de acción tanto endocrino, al modular el metabolismo sistémico (incluyendo la acción de la insulina en los adipocitos subcutáneos), como factor autocrino y paracrino, al regular la biología local del TAV9.
La omentina es una proteína de secreción formada por 313 aminoácidos y con un peso molecular de 33 KDa y es específica de depósitos de grasa. Es codificada por 2 genes (1 y 2), localizada en una región cromosomal de 1q22-q23, y estimula el consumo de glucosa mediado por la insulina en adipocitos humanos. La omentina es detectable en sangre de humanos en concentraciones de 100-800 ng/ml, y su concentración disminuye en humanos obesos y con sobrepeso, en diabetes tipo 2, síndrome metabólico, disfunción endotelial y enfermedad de la arteria coronaria9 10 11; sin embargo, su concentración se incrementa después de que el individuo pierde peso12.
Distintos estímulos regulan la secreción de insulina, por ejemplo, la interleucina-6 (IL-6) induce la secreción de omentina en las células endoteliales y las células del estroma vascular, mientras que los niveles plasmáticos de glucosa e insulina altos (resistencia a la insulina) disminuyen su secreción, lo que muestra que la actividad metabólica e inflamatoria son reguladores importantes en la actividad de la omentina, y que esta se encuentra implicada en el proceso inflamatorio13.
Inflamación en la obesidad y omentina
A medida que las personas incrementan su peso, los adipocitos se hipertrofian, debido a un aumento excesivo en sus depósitos de lípidos almacenados, lo que resulta en alteraciones moleculares y celulares que posteriormente afectan el metabolismo sistémico. En condiciones normales el adipocito se encarga de usar la glucosa captada para la producción y almacenamiento de triglicéridos (como reservorio de energía) mediante la lipogénesis. Sin embargo, cuando se presenta un exceso de nutrientes, los triglicéridos sobrecargan la capacidad de almacenamiento de los adipocitos, favoreciendo cambios en la arquitectura celular del mismo, iniciando así estrés celular14.
El estrés celular es un estado de disfunción celular, y entre otras cosas, culmina en procesos apoptóticos e inflamatorios; dicho estrés es inducido en los adipocitos por cinasas de estrés, entre ellas la cinasa c-jun N terminal (JNK). La activación de JNK es iniciada por la acumulación de diacilgliceroles (DAG), ácidos grasos saturados y ceramidas (resultado del almacenamiento excesivo de triglicéridos)14 15.
Por otro lado, la hipoxia es también resultado de la expansión e hipertrofia del TAV; y esta, junto con los DAG, provocan la producción endógena de citocinas proinflamatorias por parte del adipocito, las cuales inician, aumentan y mantienen la respuesta inflamatoria en tejido hepático, muscular y adiposo (fig. 1). La disfunción metabólica durante la obesidad resulta de la producción de las moléculas anteriormente mencionadas y se asocia con la activación de diversas vías de señalización inflamatorias. El aumento de DAG provoca la activación de isoformas de proteincinasas C, entre ellas proteincinasas C λ o θ, quienes fosforilan a inhibidor del factor nuclear KB, resultando en la translocación del factor nuclear kappa B (NF-κB) hacia el núcleo, comenzando así la transcripción de moléculas proinflamatorias (IL-1β, TNFα, IL-6) y las quimiocinas (MCP-1, IL-8). Lo anterior provoca la activación de macrófagos residentes del tejido adiposo, que, ante dicho microambiente inflamatorio, comienzan a acumularse en el TAV produciendo, manteniendo y aumentando la inflamación local, a través de la secreción de TNFα, IL-1β, EROS y metaloproteinasas14 15 16. Dicha actividad inflamatoria provoca daño al endotelio y a otros tejidos relacionados durante la obesidad. El estrés del retículo endoplásmico es también un proceso activado por la lipogénesis excesiva, en donde el aumento de ceramidas y la constante estimulación de TNFα (secretada por el tejido adiposo adyacente) provoca la activación de JNK, que también inhibe la captación de glucosa y fosforila al factor de transcripción activador de proteína 1, aumentando la síntesis de moléculas proinflamatorias, al igual que incrementa la activación de proteínas apoptóticas14 15 16 (fig. 1).

Figura 1 Obesidad e inflamación. El aumento y la sobrecarga en los depósitos de triglicéridos en los adipocitos conduce a la acumulación intracelular de DAG y ceramidas, y esto favorece inflamación, estrés oxidante y estrés del retículo endoplásmico. Estas condiciones aumentan la activación de vías mediadas por JNK e inhibidor del factor nuclear KB (IKKB), estableciendo un estado de resistencia a la insulina. A su vez, estás vías convergen en la síntesis de moléculas proinflamatorias (TNFα, IL-1B) y moléculas quimioatrayentes de monocitos y neutrófilos (MCP-1 e IL-8 respectivamente). El contacto de los macrófagos residentes con estas sustancias y con moléculas liberadas por adipocitos necróticos (ATP, K+, HMGB-1), provocan su activación, aumentando el estado de resistencia a la insulina y por lo tanto, el riesgo de enfermedad cardiovascular.
Las disfunciones celulares resultantes de la respuesta inflamatoria y de estrés favorecen un aumento en la lipólisis con la subsecuente liberación de ácidos grasos libres, los cuales activan a los macrófagos a través de receptores tipo Toll, involucrados en el inicio de la respuesta inflamatoria innata, comenzando un ciclo de retroalimentación positiva, pues las citocinas liberadas por los macrófagos activados mantienen y amplifican la respuesta inflamatoria en los adipocitos y células endoteliales. Dicha inflamación determina la disfunción en la actividad metabólica caracterizada por resistencia a la insulina, perpetuando aún más la respuesta inflamatoria14 15 (fig. 1). Todo esto induce disfunciones metabólicas e inflamatorias sistémicas, y el comienzo de aterosclerosis y disfunción endotelial. En comparación con las personas delgadas, el tejido adiposo en los individuos obesos muestra una mayor expresión de proteínas proinflamatorias, incluyendo el TNFα e IL-62.
La omentina es capaz de disminuir la actividad inflamatoria que ejerce TNFα e IL1-β, a través de la inhibición de JNK y NF-κB17. También, a través de la β-oxidación, evita la formación de DAG y ceramidas, disminuyendo así el comienzo de la inflamación mediada por los adipocitos hipertróficos14 15 16 17. Estos hallazgos muestran que la omentina puede participar en la respuesta inmune innata inducida durante la obesidad, y puede tener efectos cardiovasculares y metabólicos benéficos, ejerciendo un papel antiinflamatorio importante en los estados de inflamación crónica, como la obesidad y la diabetes, evitando así el comienzo de la aterosclerosis, disminuyendo la disfunción endotelial9. Debido a que la omentina se expresa predominantemente en las células vasculares estromales del TAV, esta puede potenciar el papel antiinflamatorio y ser crítica en la regulación de los mediadores inflamatorios del tejido adiposo, especialmente para los macrófagos residentes del TAV, los cuales son uno de los principales componentes de la fracción del estroma vascular y desempeñan un papel crítico en la iniciación, mantenimiento y la resolución de la inflamación, así como en el comienzo de las lesiones aterogénicas12.
Omentina en la resistencia a la insulina y el metabolismo energético
La homeostasis energética es el proceso por el cual el cuerpo establece un nivel de energía constante, que coincide con el equilibrio entre el consumo y el gasto energético, además de almacenar energía para necesidades futuras. La insulina es un regulador importante en este proceso. Una de las funciones más importantes de la insulina es aumentar la captación de glucosa en las células especializadas en la función metabólica (hepatocitos, miocitos y adipocitos) para su posterior oxidación o almacenamiento. Este proceso es estimulado por la activación de una serie de cascadas que involucran la fosforilación de enzimas río abajo18. En condiciones normales, la insulina se secreta como resultado del aumento en los niveles de glucosa; una vez secretada se une al receptor de la insulina (IR) en la superficie celular de los tejidos metabólicos antes mencionados. Todas las respuestas iniciadas mediante la unión de la insulina a su receptor están mediadas por la activación de vías de transducción de señales19 20.
La activación del IR provoca la autofosforilación de sus dominios tirosincinasa, lo que resulta en la asociación de sustratos del receptor de insulina (de los cuales hay 4: IRS1, IRS2, IRS3 e IRS4) con el receptor. Lo anterior resulta en la activación de la fosfatidilinositol-3-cinasa) que, a su vez, activa AKT (o proteincinasa B) mediante su fosforilación. Tras su activación, AKT media la fosforilación y activación de la glucógeno sintetasa cinasa-3 y a la GTPasa RAB 10, la cual facilita la translocación del transportador de glucosa 4 sobre la superficie celular (en adipocitos y miocitos) para la captación y el transporte de glucosa hacia las células. Esto se traduce en un aumento en la captación y metabolismo de la glucosa19 20 (fig. 2).
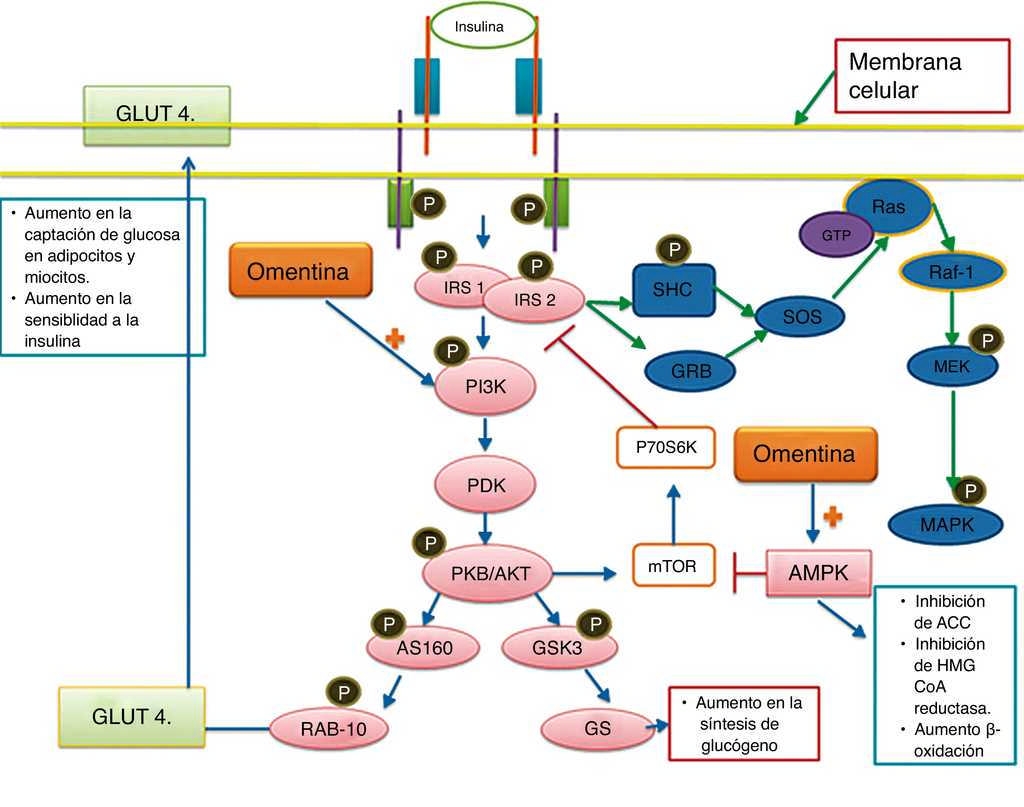
Figura 2 Vía de señalización de la insulina y omentina. El receptor de la insulina (IR) es un receptor tirosincinasa heterotetrámero, su activación facilita la translocación del transportador de glucosa 4 (GLUT 4) a la superficie celular de adipocitos y células musculares, favoreciendo así la captación de glucosa y la glucogenogénesis. La fosforilación de IRS también conlleva la activación las MAPK. La omentina estimula la captación de glucosa, además de la síntesis de glucógeno, también estimula la activación de AMPK que disminuye la resistencia a la insulina, la síntesis de triglicéridos y colesterol, y favorece la β-oxidación; y a la 3-hidroxi-3-metilglutaril-CoA (HMG-CoA) reductasa, disminuyendo así la síntesis de colesterol en el hígado.
La sensibilidad de las células a la insulina se encuentra finamente regulada y los IRS son de las enzimas más importantes en este sistema; estos contienen varios posibles sitios de fosforilación de tirosina y aproximadamente 50 sitios potenciales de fosforilación serina/treonina20 21. La fosforilación en los residuos tirosina favorece la activación de las actividades de captación, metabolismo y almacenamiento de la glucosa, mientras que la fosforilación en los residuos serina involucra la inhibición de la señalización metabólica del IR; dichas actividades pueden ser reguladas por retroalimentación positiva y negativa19 20. Por ejemplo, AKT puede activar la vía que conduce a mTOR (objetivo de la rapamicina en mamíferos) y mTOR-p70S6K propicia la fosforilación de serina, disminuyendo la actividad del IRS19 20.
Los mediadores inflamatorios y de estrés (inhibidor del factor nuclear KB y JNK respectivamente) en los adipocitos son capaces de alterar la función del IR al fosforilar la porción serina en el IRS1 y 2, inhibiendo así la vía metabólica IP3K/AKT, resultando en un estado de resistencia a la insulina14 15. En los adipocitos, la omentina incrementa la fosforilación de AKT probablemente a través de Loc40139721, y por lo tanto, aumenta el transporte de glucosa mediado por la insulina, lo que sugiere un papel importante en el metabolismo de la glucosa y la regulación de la sensibilidad a la insulina. La omentina también es capaz de disminuir la actividad inflamatoria mediada por NF-κB, y mediante su inhibición (al igual que a través de la inhibición de JNK) muestra que la omentina ejerce también un aumento en la sensibilidad a la insulina en adipocitos, miocitos y hepatocitos a través de su efecto antiinflamatorio17. Sin embargo, el papel de la omentina en la inhibición de NF-κB y JNK ha sido demostrado en el endotelio, por lo que es necesario dilucidar este efecto en adipocitos, pues implicaría un posible efecto en la regulación del síndrome metabólico.
Por otro lado, la omentina aumenta la actividad de IRS mediante la inhibición de la vía mTOR-p70S6K a través de la activación de otro regulador importante en la homeostasis metabólica, la proteína cinasa activada por AMP (AMPK). La AMPK actúa como un sensor metabólico del estado de energía celular18 y es activada por la fosforilación de su residuo treonina; su activación inhibe a las enzimas involucradas en la síntesis del colesterol y ácidos grasos: 3-hidroxi-3-metilglutaril-CoA reductasa y acetil CoA carboxilasa (fig. 2). La activación de AMPK promueve la β-oxidación de ácidos grasos y la captación de glucosa en los miocitos, reduce la gluconeogénesis en los hepatocitos, e inhibe la lipogénesis en adipocitos, lo que muestra un papel importante de la omentina en el metabolismo energético y en la disminución de la sobrecarga de lípidos21. Esto apoya el uso, en potencia, de la omentina en pacientes con síndrome metabólico, pues el aumento del TAV se encuentra asociado a la acumulación de triglicéridos en el músculo e hígado, contribuyendo al inicio de síndrome metabólico y resistencia a la insulina, lo cual aumenta el riesgo de eventos cardiovasculares15 19. También, la inadecuada activación del IR provoca un aumento en los niveles de ácidos grasos libres, como resultado de la constante lipólisis de triglicéridos en los adipocitos y de la estimulación por citocinas proinflamatorias. Los ácidos grasos libres favorecen la producción elevada de lipoproteínas de muy baja densidad en el hígado, y con esto, un incremento en la formación de lipoproteínas de baja densidad (LDL), aumentando así el riesgo cardiovascular; a su vez, activan a los macrófagos residentes del tejido adiposo (a través de receptores tipo Toll), los cuales exacerban la resistencia a la insulina mediante la producción de más citocinas inflamatorias15 16 (fig. 3), procesos en los cuales la omentina podría implicar una regulación, por los mecanismos antiinflamatorios antes descritos.
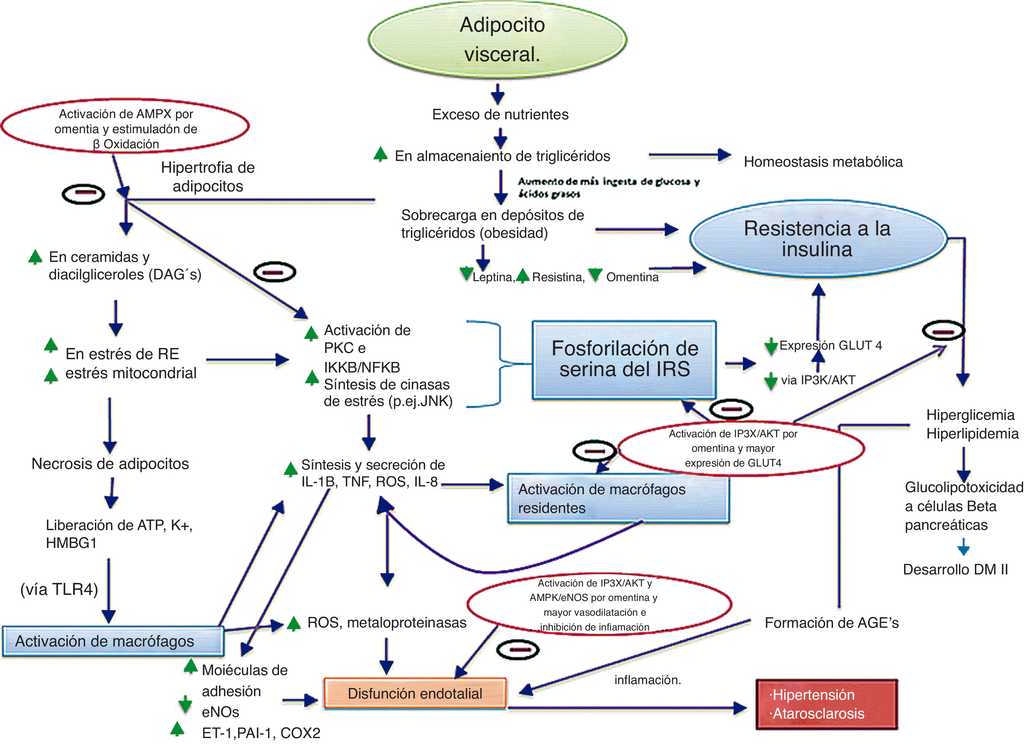
Figura 3 Mecanismos de resistencia a la insulina mediados por inflamación y disfunción endotelial resultante y los efectos de la omentina en estas vías. La acumulación de DAG en los adipocitos favorece la activación de isoformas proteincinasas C (PKC) y JNK, aumentando la inflamación y culminando en un estado de resistencia a la insulina. Los macrófagos residentes son también activados por adipocitos hipertróficos y por moléculas de adipocitos en necrosis, favoreciendo la expresión de E-selectina e ICAM-1 en el endotelio, contribuyendo a la adhesión celular. La hiperglucemia favorece la formación de AGE, los cuales aumentan la disfunción endotelial. La omentina impide la resistencia a la insulina a través de la fosforilación de IP3K/AKT, y aumenta la β-oxidación a través de la activación de AMPK. La omentina inhibe, además, al factor NF-κB, disminuyendo así la expresión de citocinas proinflamatorias.
Experimentos in vitro muestran que el tratamiento con omentina recombinante aumenta el consumo de glucosa mediado por la insulina en adipocitos viscerales y subcutáneos de humanos, mediante el incremento en la fosforilación de AKT21. Por lo tanto, la omentina puede desempeñar una función paracrina o endocrina en la modulación de la sensibilidad de la insulina y el metabolismo de la glucosa en la grasa visceral. Adicionalmente, la circulación sanguínea de omentina puede ejercer, en otros órganos como el músculo, hígado y grasa subcutánea, un aumento en la sensibilidad a la insulina y el metabolismo de la glucosa22 (fig. 3).
En el plasma humano los niveles de omentina están disminuidos en la diabetes mellitus tipo 2 y tipo 123 24, lo que muestra que la omentina ejerce un papel importante en el estado de resistencia a la insulina en los adipocitos subcutáneos y viscerales e infiere una relación entre la omentina y los niveles de glucosa e insulina22. La metformina es el primer fármaco de sensibilización a la insulina en demostrar que su uso incrementa los niveles de omentina en mujeres con sobrepeso, resistentes a insulina y con síndrome de ovario poliquístico25 26 27. En otro estudio, el tratamiento combinado de metformina y liraglutida (un péptido-1 similar al glucagón humano) en pacientes con diabetes no controlada produjo un aumento de los niveles de omentina después de un tratamiento de 6 meses28. Tan et al. también confirman que la omentina decrece en pacientes femeninas con síndrome de ovario poliquístico, el cual está asociado a la resistencia a la insulina y la obesidad, pero los niveles se incrementan después del tratamiento con metformina13. Esto sugiere el uso de metformina como inductor de omentina y sus efectos benéficos en la resistencia a la insulina.
Disfunción endotelial y omentina
Recientemente, un estudio demostró que la inflamación del tejido adiposo ejerce distintos efectos sobre la vasculatura a través de la presencia de macrófagos dentro del TAV que expresan y liberan citocinas. Estas citocinas alcanzan el hígado a través de la circulación portal, produciendo así la inflamación hepática, además de inducir una respuesta inflamatoria sistémica y crónica a través de la activación del endotelio29.
La resistencia a la insulina debida a la inflamación sistémica de bajo grado culmina en diversos mecanismos de daño endotelial. Entre ellos, se encuentra el daño mediado por la acumulación de productos finales de glucosilación avanzada (AGE), como consecuencia de un estado crónico de hiperglucemia (debido a la resistencia a la insulina)30 31.
Los AGE provocan la activación de los receptores RAGE en endotelio y macrófagos, favoreciendo así la disfunción endotelial a través del aumento en la producción de ROS y metaloproteinasas, y de la secreción de citocinas proinflamatorias (IL-1B, TNFα), por parte de macrófagos; y de moléculas activadoras de plaquetas y anti-fibrinolíticas (APF e inhibidor del activador del plasminógeno-1 respectivamente) y sustancias vasoconstrictoras (tromboxanos, endotelina), por parte del endotelio (fig. 3), lo que finalmente provocan daño al glucocáliz del endotelio32.
Además, la activación del endotelio por TNFα e IL-1β estimula la expresión de selectinas (E-selectina) y moléculas de adhesión intercelular (ICAM-1) involucradas en la atracción de neutrófilos y monocitos, que exacerban el daño inflamatorio crónico, estableciendo un estado proaterogénico y protrombótico (fig. 3). El daño al glucocáliz resulta en el crecimiento de la permeabilidad del endotelio, con un aumento en la filtración de LDL al espacio subendotelial, provocando un incremento en la actividad inflamatoria mediada por macrófagos residentes y el subsecuente inicio de la aterosclerosis31 32 33. La oxidación de las moléculas LDL es llevada a cabo por macrófagos, los cuales, mediante receptores fagocíticos las captan a su interior donde son oxidadas por la acción de la mieloperoxidasa, provocando la diferenciación de macrófagos en células espumosas. Las células espumosas son las iniciadoras de la lesión aterogénica, pues los productos de secreción de estas provocan inflamación endotelial y quimioatracción de neutrófilos y monocitos (a través de IL-8 y MCP-1 respectivamente), y estos, mediante la producción de ROS y metaloproteinasas, median la degradación de la matriz extracelular, además de la estimulación en la migración de las células musculares lisas, iniciando así la formación de la placa aterogénica, conformada por los componentes antes mencionados, además de macrófagos necróticos, que son parte del núcleo de la placa, estableciendo un estado proinflamatorio y protrombótico33 34. Se conocen los efectos que la omentina ejerce en la actividad inflamatoria, sin embargo, aún no hay estudios que impliquen su papel en la protección contra el desarrollo de la placa ateromatosa.
En situaciones normales, las moléculas encargadas de la regresión en la lesión aterogénica son las HDL, que propician y favorecen el transporte inverso del colesterol, mediante el eflujo de colesterol del interior de los macrófagos en la capa subíntima de las arterias a través de la activación del receptor ABCA 1 (entre otros). Finalmente, la eliminación del colesterol es llevada a cabo en el hígado, pero durante el estado de resistencia a la insulina y la obesidad estas moléculas antiaterogénicas se encuentran disminuidas (dislipidemia), reduciendo así los mecanismos encargados en la prevención del daño34 35.
El daño a los vasos, con la subsecuente disfunción endotelial, puede también ser resultado de la pérdida del equilibrio entre la producción de óxido nítrico (NO) vasodilatador y endotelina (ET-1) vasoconstrictora, como consecuencia de la inhibición de la vía metabólica IP3K/AKT en el endotelio (dependiente de la fosforilación tirosina en el IRS en el endotelio). Como se mencionó antes, esta vía es inhibida por los mediadores inflamatorios producidos por adipocitos, macrógafos y células endoteliales, que fosforilan la porción serina del IRS, resultando así, en una menor actividad en la vía IP3/AKT, que resulta, además de la disminución en la expresión de transportador de glucosa 4 en la superficie celular, en la inhibición en la fosforilación de óxido nítrico sintasa (eNOS) y la subsecuente disminución en la síntesis de NO vasodilatador (fig. 4). Además, esto favorece la producción de ROS, aumentando el daño al endotelio35. Por otro lado, la señalización de la insulina no solo activa la vía metabólica AKT, sino que involucra también la activación de la vía de crecimiento y de las proteincinasas activadas por mitógenos (MAPK); esta vía es responsable de la producción de ET-1 en el endotelio mediante la activación de la MAP cinasa ERK (cinasa regulada por señales extracelulares). La vía MAPK/ERK no es afectada por los mediadores inflamatorios, con lo que en un estado de hiperinsulinemia e inflamación (síndrome metabólico) la insulina sobrecarga y activa esta vía. Lo anterior provoca la inclinación de la balanza hacia la producción de ET-1 y por lo tanto la subsecuente disfunción endotelial con predominio de actividad vasoconstrictora33 (fig. 4). Diversos reportes indican que la omentina ha sido implicada en muchas enfermedades crónicas de tipo inflamatorio36 y se ha reportado que la omentina induce vasorrelajación en el endotelio mediado por la activación de la vía del NO sugiriendo que la omentina puede participar en la funcionalidad adecuada del endotelio, y que puede estar implicada en la enfermedad cardiovascular mediante la modulación de la reactividad contráctil e inflamatoria de los vasos sanguíneos37.

Figura 4 Disfunción endotelial asociada a inflamación y a resistencia a la insulina. Los adipocitos inflamados secretan TNFα, y conllevan la activación de macrófagos M1, los cuales a su vez provocan más inflamación en los adipocitos. La inflamación local se convierte en inflamación sistémica, en la que los productos de inflamación ejercen activación y subsecuente disfunción del endotelio. Mediante la unión de TNFα a TNFR1 se activan vías de señalización celular que convergen en genes proinflamatorios, aumentando quimioatracción, adhesión, síntesis de ROS y daño en el endotelio. Esto conduce a la inhibición y disminución en la síntesis de NO. Por otro lado, la hiperinsulinemia favorece la disfunción endotelial, debido a la constante síntesis de ET-1. La omentina disminuye estos mecanismos mediante la activación de IP3K/AKT y de AMPK, e inhibe a JNK y NF-κB lo que condiciona una menor expresión de ICAM-1, así como de COX-2 e interleucinas proinflamatorias, condicionando una actividad antiinflamatoria. La omentina también tiene efectos de reparación y angiogénesis mediante VEGF (no mostrado en la figura).
También, la inflamación vascular, al igual que el estrés oxidante asociado, propician una mayor generación de los productos derivados del ácido araquidónico; esto favorece un cambio de vasodilatación y antitrombosis en el endotelio a vasoconstricción, protrombosis e inflamación como resultado de la activación de COX-238. La omentina también atenúa la expresión de la ciclooxigenasa-2 (que es inducida en sitios de inflamación) y la activación de JNK en células endoteliales estimuladas por citocinas (p. ej., TNFα) a través de la activación de la vía AMPK/eNOS/NO posiblemente a través de Loc40139739 40 41. Mediante la inhibición de JNK, P38, NF-κB y ERK, a través de la activación de AMPK, la omentina suprime la expresión de las moléculas de adhesión en la superficie celular de células endoteliales y el estrés oxidante (ICAM, NADPHO y COX-2). Por lo tanto, es probable que las funciones de la omentina como un modulador negativo de la inflamación vascular sean importantes. En conjunto, estos datos sugieren que la ausencia de omentina representa un biomarcador de riesgo cardiovascular, y es una adipocina responsable de la modulación de la patogénesis de trastornos vasculares2 37 39 40 41 42 43 (fig. 4). Por otro lado, se demostró que la omentina inhibe la diferenciación osteoblástica de las células musculares lisas vasculares mediante la ruta de señalización de fosfatidilinositol-3-cinasa/AKT, confiriendo protección vascular42.
Omentina en la enfermedad de las arterias coronarias
La omentina se encuentra abundantemente expresada en el tejido adiposo epicárdico, el cual es un depósito de grasa visceral localizado alrededor del corazón y las arterias coronarias. Debido a que el tejido adiposo epicárdico no se encuentra separado del miocardio por fascias, los factores secretados por dicho tejido pueden afectar la función contráctil cardiaca; tales factores son inducidos como resultado de la respuesta inflamatoria crónica y sistémica en respuesta al estado de resistencia a la insulina que, aunado a la disfunción endotelial, propician el aumento en el riesgo cardiovascular43.
La disminución de los niveles de omentina se asocia significativamente con un mayor riesgo para la enfermedad de la arteria coronaria en mujeres posmenopáusicas, al igual que en varones. Al comparar los niveles de omentina de pacientes diabéticos y pacientes no diabéticos, se reportó que la omentina fue significativamente menor en los diabéticos44 45.
Se ha puesto de manifiesto el efecto promotor de la vasodilatación de la omentina en células endoteliales, mostrando que la omentina actúa como un modulador de la función vascular46. Además, también se ha demostrado que la omentina suprime la expresión ICAM-1 en las células endoteliales39, mediante la inhibición de las vías activadas por TNF e IL1-β/NF-κB y de MAPK (ERK, JNK), disminuyendo así la quimiotaxis de neutrófilos y monocitos inflamatorios. Lo anterior también disminuye la expresión de fuentes de radicales libres tales como la COX-2, confiriendo protección vascular38 (fig. 4).
Debido a todos estos eventos, la disminución de la omentina podría estar involucrada en el desarrollo de enfermedad de la arteria coronaria a través de la correlación entre los niveles bajos de omentina y la disminución en la vasodilatación coronaria dependiente del endotelio, la actividad de la insulina y la inflamación47.
Con base en las evidencias mencionadas, la omentina puede ser un fármaco implicado en la regresión de la placa aterogénica, al igual que un fármaco antitrombótico, por lo que deberían realizarse investigaciones enfocadas en este escenario de cardioprotección.
Omentina y revascularización
AKT y eNOS son reguladores cruciales del crecimiento de los vasos sanguíneos y de la función vascular de la célula. La omentina promueve la diferenciación celular endotelial y la supervivencia mediante la fosforilación de AKT/eNOS en las células endoteliales44. Además, la omentina promueve la revascularización en el músculo isquémico en ratones in vivo, lo cual está asociado con el aumento de la fosforilación de AKT/eNOS. Es de destacar que el impacto de la omentina en el reclutamiento de los vasos sanguíneos fue suprimido en ratones eNOS-KO, por lo que estos datos muestran que el eje regulador omentina-AKT-eNOS puede promover la función de las células endoteliales en condiciones de isquemia, acelerando así el proceso de revascularización46 48.
Estos estudios muestran también que la omentina promueve la ruta de señalización de AMPK en las células endoteliales y en tejido isquémico. La inhibición en la activación de AMPK fue evitada por la estimulación inducida por la omentina en la activación de AKT en la diferenciación y supervivencia de las células endoteliales in vitro. Por lo tanto, la omentina puede promover la señalización de AKT y subsecuentemente la respuesta de las células endoteliales vía AMPK48.
Se ha demostrado que la eNOS es protectora contra diversas enfermedades vasculares, incluyendo la aterosclerosis. La omentina promueve la vasodilatación en la aorta aislada de la rata46. Un reporte previo muestra que la omentina reduce la respuesta inflamatoria inducida por las citocinas en cultivos de células endoteliales, mediante la activación de eNOS. De acuerdo con estos hallazgos, el estudio muestra que la omentina aumentó la activación de la fosforilación de la eNOS por parte de la activación de AMPK en el músculo isquémico y en cultivo de células endoteliales. Estos datos también indican que las acciones de la omentina sobre el comportamiento de las células endoteliales y el crecimiento de vasos es mediado, al menos en parte, a través de su capacidad para activar AMPK/eNOS17.
Estas observaciones muestran que la eNOS actúa como un mediador fundamental de las acciones de protección de omentina sobre la vasculatura. Por otro lado, el VEGF es un factor de crecimiento angiogénico crucial que estimula la vía de señalización de AKT-eNOS en las células endoteliales, promoviendo así la revascularización y vasodilatación in vivo. Se ha demostrado que la omentina aumenta los niveles circulantes de VEGF después la cirugía isquémica47, por lo tanto, además de las acciones directas sobre células endoteliales, la omentina puede estimular la revascularización inducida por isquemia, por el aumento de la producción de VEGF. También se ha demostrado que la falta del receptor TNFα (TNFR2/p75) causa la recuperación del flujo sanguíneo deficiente y una clara reducción de la expresión del VEGF en el músculo isquémico49. Aunque la capacidad de la omentina para aumentar la producción de VEGF puede ser mediada, al menos en parte, a través de TNFR2/p75, se requiere investigación futura para dilucidar la posible relación entre omentina y la señalización del receptor de TNF.
Conclusiones
La omentina es una adipocina secretada por el TAV y está implicada en la regulación del metabolismo de la glucosa y la β-oxidación de lípidos en los principales tejidos metabólicos (hígado, tejido adiposo y músculo), al igual que la disminución de procesos inflamatorios en el endotelio. Esto muestra un rol importante contra el desarrollo de la resistencia a la insulina y el estado proinflamatorio que determina la obesidad y que culmina en diabetes mellitus tipo 2 y aterosclerosis. La omentina ofrece protección al endotelio mediante la expresión de AMPK/eNOs, estimulando una correcta actividad vasodilatadora, además, es capaz de mantener un estado antiinflamatorio al inhibir vías de transcripción inflamatorias que culminan en la expresión de moléculas de adhesión y protrombóticas. La omentina tiene también la capacidad de aumentar los niveles de VEGF, confiriendo un estado constante de angiogénesis, y por lo tanto, protección y regeneración ante el ambiente proinflamatorio (p. ej., ROS, metaloproteinasas, etc.) (fig. 5). Por lo anterior, es importante la consideración, mediante el desarrollo de estudios clínicos, de la omentina como una posible línea de tratamiento para el síndrome metabólico, al igual que para aterosclerosis. Si bien la omentina ejerce antiinflamación en el endotelio, se debería investigar más acerca de sus efectos antiinflamatorios en los adipocitos, donde recae la base y el inicio de la resistencia a la insulina.

Figura 5 Resumen: funciones de la omentina. La omentina ejerce funciones metabólicas e inmunes importantes que resultan en efectos cardioprotectores, su uso debe seguir en estudio para el tratamiento de enfermedades crónicas inflamatorias como la obesidad y la diabetes mellitus tipo 2, de la cual resultan los padecimientos cardiovasculares.
Aún más, tienen gran interés las vías de señalización que son estimuladas por la omentina, puesto que ejercen un efecto sensibilizador a la insulina, favoreciendo así la captación y procesamiento de la glucosa. Esto puede evitar el acúmulo de AGE en el endotelio, con la subsecuente activación e inflamación del mismo. Mediante la estimulación de la AMPK y la subsecuente β-oxidación de lípidos y la glucólisis, se evita la acumulación excesiva de nutrientes dentro de las células, además de disminuir las cifras de LDL (y aumentar las de HDL) en sangre, los cuales, son procesos clave en la activación de cascadas inflamatorias y de estrés, y por lo tanto, en el establecimiento la resistencia a la insulina, síndrome metabólico y aterogénesis.
Todo lo anterior confiere interés en realizar más estudios implicados en la actividad antiinflamatoria y metabólica de la omentina a nivel cardiovascular, puesto que tiene un efecto importante en la modulación de procesos fisiopatológicos que culminan en daño vascular; esto podría mostrar que la omentina puede ser un fármaco potencial contra la enfermedad cardiovascular relacionada con la obesidad y/o la diabetes mellitus tipo 2.

