











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los tumores cardiacos primarios son muy poco frecuentes en todas las edades, su prevalencia en la población general es del 0.002%1. En la edad pediátrica se ha informado que la prevalencia es del 0.027 al 0.08% en autopsias, y de hasta un 0.3% en estudios ecocardiográficos; se ha detectado hasta un 0.14% en estudios fetales, con una prevalencia de 1:10,0002-4. La mayoría de estos tumores son benignos (90%). Histológicamente los tumores benignos más frecuentes en etapa pediátrica son los rabdomiomas (60%), compuestos por las características células «en araña» (Fig. 1), seguidos de los fibromas (12%), los mixomas (10%), los teratomas intracardiacos (25%) y los hemangiomas5.

Figura 1 Histología de las células del rabdomioma. Característica «célula en araña» con núcleo desplazado, vacuolada, con granulaciones en el citoplasma y delgadas extensiones del citoplasma (cortesía del Dr. Alberto Aranda, médico patólogo del Instituto Nacional de Cardiología).
El rabdomioma lo describió von Recklinghausen en 1862 asociado a neurofibromatosis6,7. Este tumor suele estar circunscrito, lobulado, de color blanquecino o grisáceo, y hasta en el 90% de los casos es múltiple. Puede afectar cualquier punto del músculo cardiaco con predominio ventricular, sobre todo ventrículo izquierdo, pudiendo comprometer la función ventricular, la función valvular u ocasionar obstrucción del flujo8,9. Los tumores que se encuentran en las aurículas pueden comprimir las arterias coronarias ocasionando isquemia miocárdica9.
Los rabdomiomas comienzan a aparecer entre la semana 20 a 30 de gestación, el diagnóstico más temprano se ha hecho en la semana 15. Se ha visto que el crecimiento es lento a partir de las 32 semanas hasta el fin del embarazo y se detiene después del nacimiento, este comportamiento puede estar relacionado con las hormonas del embarazo9,10.
La ecocardiografía bidireccional es un estudio no invasivo y ha demostrado ser el método diagnóstico preferido, y gracias al aumento en su disponibilidad se ha incrementado la detección de estos tumores1. (Fig. 2) La resonancia magnética (RM) y la tomografía computarizada multidetector (TCMD) han permitido una mejor evaluación de estas lesiones11.

Figura 2 Imagen de cuatro cámaras con rabdomioma gigante en el ventrículo derecho por ecocardiograma.
La mortalidad descrita es variable, siendo los factores de riesgo más importantes para mal resultado perinatal el tamaño (> 20 mm), arritmias e hidrops10.
La mayoría de los pacientes con rabdomioma no requieren manejo quirúrgico, ya que la mayoría involuciona cerca de los 6 años de edad4,12,13. El tratamiento que se brinde dependerá de la repercusión hemodinámica que cause el tumor y de la presencia de arritmias graves; esto mismo determinará el pronóstico10.
La esclerosis tuberosa (ET), también conocida como enfermedad de Bourneville, es una enfermedad multisistémica poco frecuente con una prevalencia de 1:10,000-50,000 en la población general y en niños 1:6,80010,13. La asociación con rabdomioma cardiaco se encuentra en entre un 50 y un 70%. Cuando un tumor cardiaco aparece en el contexto de un síndrome, como la ET, la presentación es diferentes de la de aquellos que se presentan esporádicamente. La ET se caracteriza por la presencia de hamartomas en múltiples órganos, con predominio cerebral con la presencia de tuberosidades corticales y nódulos subependimarios, angiomiolipomas renales, hamartomas retinianos, fibromas subungulares y nevos epidérmicos, entre otros, siendo las lesiones cutáneas hipomelanóticas y las lesiones cerebrales tuberosas corticales las únicas manifestaciones en la etapa neonatal4,14.
En el 90% de los casos se presenta por mutaciones de novo, detectándose en la mitad de los casos la mutación en el gen TSC1/ hamartina (9q34.3 locus: 605284) y la otra mitad en el gen TSC2/tuberina (12q15 locus: 147570 y 16p 13.3 locus: 191092). Su forma de herencia es autosómica dominante con una variedad amplia de expresión, siendo su tendencia familiar hasta un 50%9,10,12.
Objetivo
Nuestro objetivo es dar a conocer nuestra experiencia a lo largo de 39 años en pacientes pediátricos con este diagnóstico dentro de nuestra institución, así como enfatizar la importancia de su detección, estudio y control por la asociación que tiene con ET y las repercusiones que esta tiene a largo plazo.
Material y método
Realizamos un estudio retrospectivo y descriptivo en el que revisamos expedientes digitales, físicos y en negativo, abarcando un periodo de enero de 1980 al 31 de marzo del 2018. Para seleccionar a nuestra población de estudio utilizamos los siguientes criterios:
Recolectamos información con respecto a la edad de diagnóstico, sexo, dato inicial que presentó el paciente, hallazgos clínicos asociados de alguna otra cardiopatía, manifestaciones clínicas, los hallazgos en ecocardiografía transtorácica (ECOTT), transesofágica, tomografía axial computarizada (TC) y/o RM, revisando en estos la localización, clasificación y número de tumores, así como la evolución y manejo que se le dio a estos, si fue quirúrgico o conservador. Por último se realizó una llamada telefónica de seguimiento para conocer su evolución y estado actual.
En cuanto al análisis estadístico, toda la información se recopiló en una base de datos en la cual se valoraron porcentajes, medias, mínimas y máximos.
Resultados
Se encontraron un total de 51 pacientes con diagnóstico de tumor cardiaco, de los cuales 24 (47%) eran rabdomiomas.
De estos pacientes, no se encontró diferencia de presentación en cuanto al sexo (12 mujeres y 12 hombres). El diagnóstico se hizo prenatal en 8 pacientes (33%), 1 por arritmia neonatal y extrasistolia ventricular y 7 como hallazgo incidental durante ultrasonido obstétrico, en 5 pacientes (21%) al nacimiento y en 11 casos (46%) durante el primer año de vida.
Las manifestaciones clínicas más frecuentes se presentan en la tabla 1.
Tabla 1 Manifestaciones clínicas presentadas. Se muestran los síntomas y signos más comunes que presentaron los pacientes. En cuanto a las arritmias, se describe cuales fueron los que presentaron alguna y su tratamiento en su mayoría fue con digitálicos y un marcapasos definitivo para el BAV completo
| Paciente | Soplo | Arritmia | Cianosis/disnea | Asintomático |
|---|---|---|---|---|
| 1 | Extrasístole ventricular | |||
| 2 | BAV 3° | |||
| 3 | x | |||
| 4 | Extrasístole ventricular y taquicardia supraventricular | |||
| 5 | Extrasístole ventricular y taquicardia supraventricular | |||
| 6 | Arritmia neonatal | |||
| 7 | x | |||
| 8 | BIRDHH | x | ||
| 9 | x | |||
| 10 | x | |||
| 11 | x | BIRDHH | ||
| 12 | Taquicardia supraventricular | |||
| 13 | x | |||
| 14 | x | |||
| 15 | x | |||
| 16 | x | |||
| 17 | x | Taquicardia supraventricular | ||
| 18 | x | x | ||
| 19 | Taquicardia supraventricular | |||
| 20 | x | |||
| 21 | x | |||
| 22 | x | x | ||
| 23 | x | |||
| 24 | Extrasístole auricular y ventricular |
BAV: bloqueo auriculoventricular; BIRDHH: bloqueo incompleto de la rama derecha del Haz de His.
El estudio de imagen que se utilizó para confirmar el diagnóstico del tumor fue la ECOTT (Fig. 3). En 5 pacientes se confirmó por estudio histopatológico.
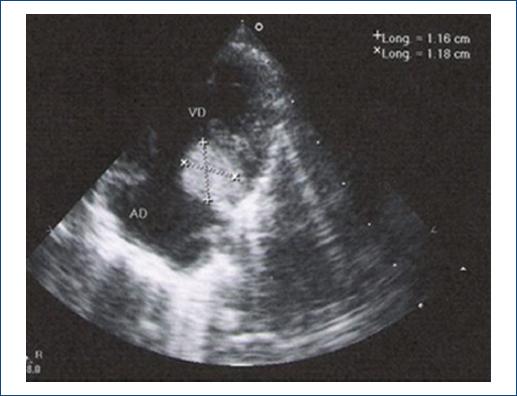
Figura 3 Imagen ecocardiográfica mostrando un engrosamiento en proyección posterior el cual corresponde a un rabdomioma.
La localización de los tumores se muestra en la tabla 2.
Tabla 2 Localización de rabdomiomas. En su mayoría, 19 casos (79%), los tumores fueron múltiples y en 5 (21%) se presentó como tumor único (estos se muestran resaltados)
| Paciente | VD | VI | VD y VI | Septum IV |
|---|---|---|---|---|
| 1 | x | |||
| 2 | x | |||
| 3 | x | |||
| 4 | x | x | ||
| 5 | x | |||
| 6 | x | x | ||
| 7 | x | x | ||
| 8 | x | |||
| 9 | x | |||
| 10 | x | |||
| 11 | x | |||
| 12 | x | |||
| 13 | x | |||
| 14 | x | |||
| 15 | x | |||
| 16 | x | x | ||
| 17 | x | x | ||
| 18 | x | |||
| 19 | x | |||
| 20 | x | |||
| 21 | x | |||
| 22 | x | |||
| 23 | x | |||
| 24 | x |
VD: ventrículo derecho; VI: ventrículo izquierdo.
Diez de los pacientes (41%) además de la presencia de rabdomiomas presentaron coexistencia de alguna cardiopatía congénita: 1 con estenosis pulmonar (4%), 1 con hipoplasia de aorta ascendente y arco aórtico (4%), 2 con foramen oval permeable (8%), 2 pacientes con comunicación interauricular (8%), 3 con persistencia del conducto arterioso (PCA) (12%) y un paciente con trisomía 21 que presentaba comunicación interventricular y PCA.
En 17 de ellos (71%) se hizo diagnóstico de ET, que se confirmó por imagen con TC y/o RM de encéfalo con presencia de calcificaciones interventriculares (Fig. 4).

Figura 4 Tomografía de cráneo corte transversal, donde se observan las tuberosidades intracraneanas, característico de la esclerosis tuberosa.
Las manifestaciones clínicas que presentaron los pacientes se muestran en la tabla 4.
Tabla 3 Criterios diagnósticos de esclerosis tuberosa (ET)*
| Criterios genéticos |
|---|
| La identificación de una mutación en TSC1 o TSC2 en el ADN es suficiente para hacer un diagnóstico definitivo de ET |
| Del 10 al 25% de los pacientes no tienen mutación identificada por pruebas genéticas convencionales, y un resultado normal no excluye mutación en TSC |
| Criterios clínicos |
| Criterios mayores |
| 1. Máculas hipomelanóticas (≥ 3, de al menos 5 mm de diámetro) |
| 2. Angiofibromas (≥ 3) o placa fibrosa cefálica |
| 3. Fibroma ungueal (≥ 2) |
| 4. Placas de Shagreen |
| 5. Hamartomas retinianos múltiples |
| 6. Displasias corticales |
| 7. Nódulos subependimarios |
| 8. Astrocitoma subependimario de células gigantes |
| 9. Rabdomioma cardiaco |
| 10. Linfangioleiomiomatosis |
| 11. Angiomiolipomas (≥ 2) |
| Criterios menores |
| 1. Lesiones dérmicas «en confeti» |
| 2. Hoyos en esmalte dental (≥ 3) |
| 3. Fibromas intraorales (≥ 2) |
| 4. Parche acrómico de la retina |
| 5. Quistes renales múltiples |
| 6. Hamartomas no renales |
*Diagnóstico definitivo: 2 criterios mayores o 1 criterio mayor con ≥ 2 criterios menores. Diagnóstico probable: 1 criterio mayor o ≥ 2 criterios menores.
Adaptada con permiso de Northrup, et al., 2013.9
Tabla 4 Características clínicas de esclerosis tuberosa (ET)
| Características clínicas de ET | |
|---|---|
| Serie 1 | |
| Manchas hipocrómicas | 7 |
| Crisis convulsivas | 10 |
| Hamartoma retiniano | 1 |
| Angiofibromas | 1 |
| Placas de Shagreen | 1 |
| Fibroma digital | 1 |
| Angiomiolipoma renal | 1 |
| Angiomiolipoma hepático | 1 |
| Exotropía | 1 |
| Retraso mental | 3 |
| Asintomático | 5 |
Doce pacientes (50%) tuvieron seguimiento por neurología, 10 (41%) por dermatología, 8 (33%) por oftalmología y 4 (16%) con genética en nuestra institución, el resto fue referido para seguimiento en su hospital pediátrico correspondiente.
La mitad de los pacientes se dejó en vigilancia, a 7 (29%) se les dio tratamiento médico para control de crisis convulsivas y 5 (21%) requirieron manejo quirúrgico por repercusión hemodinámica, 2 por obstrucción del tracto de salida del ventrículo izquierdo, 2 por obstrucción del tracto de salida del ventrículo derecho y uno por hipoxia. Diecisiete pacientes (71%) se mantuvieron estables, 5 (21%) presentaron regresión espontánea y 2 (8%) fallecieron, una de las pacientes por paro cardiaco debido a que presentaba bloqueo auriculoventricular completo; en este caso el rabdomioma se encontraba en el septum interventricular, lo que involucraba y afectaba el sistema de conducción. La otra paciente falleció debido a un choque cardiogénico irreversible secundario a falla ventricular posterior a la resección parcial del tumor.
Discusión
Los tumores cardiacos en la edad pediátrica son en su mayoría benignos, de ellos el rabdomioma se considera el más frecuente, sobre todo en los primeros años de vida. En la mayoría de los casos los pacientes suelen estar asintomáticos y se detectan por la presencia de soplos, pero las manifestaciones pueden variar dependiendo del sitio del tumor y las estructuras que afecte, pudiendo presentar desde un síncope, insuficiencia cardiaca, síndrome de obstrucción caval, hipertensión pulmonar, isquemia pulmonar, cerebral o miocárdica, cor pulmonale, arritmias, embolia, trombosis, hasta muerte súbita. En la vida prenatal se suele presentar con arritmias o hidrops fetal4,14.
El diagnóstico prenatal se puede hacer a partir del tercer trimestre durante el ultrasonido obstétrico habitual, detectando el tumor, hidrops o arritmias y se puede usar la ecocardiografía fetal avanzada en casos específicos de riesgo, a partir de la semana 14 de gestación. De los 8 pacientes que se detectaron prenatalmente, 7 fue como hallazgo incidental durante el ultrasonido obstétrico habitual y en 1 paciente se detectó por presentar arritmia y extrasistolia ventricular con taquicardia supraventricular.
En la etapa posnatal, el diagnóstico se hace como hallazgo en pacientes con soplo, enfermedad valvular obstructiva o en pacientes con signos y síntomas de ET o cuando hay antecedente familiar de ET, siendo diagnosticados el 80% de los casos durante el primer año de vida4,7. En nuestros pacientes el 100% de ellos se diagnosticó antes del primer año de vida, siendo el soplo cardiaco el principal motivo de estudio.
Con la ayuda del ecocardiograma podemos ver el sitio, tamaño y repercusión hemodinámica del tumor; la TCMD y la RM tienen un papel importante al ser estudios complementarios, y están indicados cuando el diagnóstico no es del todo claro o para añadir información para el plan quirúrgico, con estos podemos visualizar la localización, el tamaño, las relaciones anatómicas y compromiso de las estructuras adyacentes a la lesión, así como una mejor forma de caracterizar el tejido15,16.
En la RM el tumor se muestra isointenso en T1 e hiperintenso en T2, comparado con el miocardio y no realza con contraste, lo que permite distinguirlo del fibroma cuando es intramural. En la TCMD el tumor es hipodenso comparado al miocardio aun posterior al contraste11,12 (Fig. 5). A pesar de tener una mejor caracterización con la RM, preferimos y recomendamos el uso de la ecocardiografía debido a que la RM requiere sedación profunda que puede descompensar a un paciente crítico y su uso lo limitamos a pacientes en que el beneficio que se obtiene es mayor al riesgo en que se somete al paciente.

Figura 5 A: imagen de resonancia magnética en cine 4C de paciente de sexo masculino de 10 años de edad con aneurisma de septum interventricular en tercio medio hacia ventrículo derecho y tumor intramiocárdico en septum interventricular tercio apical. B: cine dos cámaras con tumoración intramural (9 x 6 mm) septal apical. C: T2* hipointensa. D: primer paso tumoración hipointensa intramural. E: T1 FatSat hipointensa. F: T1 hiperintensa. G: T2 W isointensa. H: imagen en realce tardío de gadolinio, hipertensa, reforzamiento tardío intramiocárdico lineal en la pared del aneurisma.
Se ha descrito involución del tumor, ya sea parcial o total, hasta en un 80% y se cree que el mecanismo por el que estos tumores presentan regresión es la apoptosis4,12,13. En nuestro estudio < 25% presentó involución (5/24), este porcentaje se explica debido a que 4/24 (16%) por las edades actuales de los pacientes aún se encuentran dentro de la ventana de tiempo para que presenten involución. En 5/24 (21%) se realizó resección quirúrgica, por lo que no logramos ver la capacidad de involución en estos pacientes. Los 8 pacientes restantes descontinuaron seguimiento en nuestra institución y no logramos establecer contacto con ellos, por lo que desconocemos la evolución de estos pacientes.
Existen reportes del uso de inhibidores de rapamicina como una alternativa a la cirugía en pacientes sintomáticos12,15. Martínez, et al. reportaron un paciente con rabdomioma gigante diagnosticado prenatalmente, quien presentó insuficiencia cardiaca y circulación sistémica dependiente de conducto después del nacimiento. Se le dio tratamiento farmacológico con everolimús, con el cual tuvo una involución de 0.80 cm2/día. Hasta el momento es el tumor más grande del ventrículo izquierdo reportado sin opción a cirugía y con involución significativa17. Hasta el 2017 no hemos tenido la necesidad de usarlo, por una parte debido a la antigüedad de algunos casos, todavía no existía el medicamento y posteriormente no estaba autorizado como tratamiento de elección, pero lo tenemos en cuenta como alternativa de tratamiento en futuros casos.
Los rabdomiomas cardiacos se ven hasta en el 50% de los pacientes con ET y pueden ser la primera manifestación fenotípica del síndrome. Cuando un rabdomioma es detectado en imagen, del 40-90% ocurre en el contexto de ET, esta asociación aumenta al 100% si se encuentra más de un rabdomioma12. De nuestros pacientes el 83% presentó ET, lo que coincide con la literatura, pero discrepamos al tener un 77% de asociación cuando hay tumores múltiples, lo que se puede deber a que al momento los pacientes no han presentado datos de ET, pero puede que al paso del tiempo tengan manifestaciones, ya que la edad que tienen es corta para descartarla, por lo que su vigilancia es de importancia.
El síntoma más común son las crisis convulsivas, las cuales pueden no responder al manejo médico, ocasionando retraso mental severo y desarrollo de hidrocefalia progresiva por las tumoraciones cerebrales. En ausencia de convulsiones la incidencia de retraso mental es bajo, pero en presencia de retraso mental las convulsiones siempre estarán presentes4. En nuestra serie, 10 de los 17 pacientes con ET presentaron crisis convulsivas, 3 de ellos con retraso mental.
Se puede sospechar la presencia de ET con la tríada de Vogt: retraso mental, convulsiones y lesiones cutáneas. Existen criterios establecidos desde 1992 para su diagnóstico (Tabla 3). La RM aporta información adicional, ayudando a detectar lesiones tuberosas cerebrales asociadas a ET y a tumores extracardiacos10. Hay que tomar en cuenta que la ausencia de lesiones cerebrales o renales en la etapa prenatal no excluye la aparición posnatal4,14.
Debido a la elevada asociación de rabdomioma con ET, es de importancia el manejo multidisciplinario con neurología para el tratamiento de estos pacientes y acudir a rehabilitación temprana para disminuir las secuelas, así como el asesoramiento genético con los padres.
Las cardiopatías congénitas con tumor cardiaco pueden coexistir, se han reportado la asociación de rabdomiomas con anomalía de Ebstein e hipoplasia de la válvula pulmonar, aunque estas asociaciones no están bien estudiadas, consideramos que hasta el momento pueden ser relaciones aisladas18. En nuestra serie encontramos una asociación en 10 de los casos. Algunos autores postulan que la presencia del tumor en etapas tempranas del desarrollo embriológico tendría una posible interacción con el crecimiento de las estructuras normales del corazón16,19.
Así mismo, la asociación de ET y aneurismas de la aorta abdominal se ha reportado en la literatura desde 1971 en pacientes en edad escolar. En el Instituto Nacional de Cardiología se reportó un caso de un paciente de 8 meses de edad con rabdomiomas múltiples, ET y aneurisma gigante de la aorta abdominal, comprometiendo desde el diafragma hasta las arterias iliacas20.
Conclusión
El rabdomioma es un tumor de histología benigna poco frecuente, pero cuando tiene repercusión importante su evolución puede ser maligna y su asociación con ET ensombrece el pronóstico.
Recomendamos que en todos los pacientes con ET debe realizarse un ecocardiograma para descartar rabdomiomas y paralelamente un electrocardiograma para descartar arritmias.
Cuando se corrobora el diagnóstico de rabdomioma, recomendamos mantener la vigilancia de la regresión espontánea; pese a ello es de importancia mantener una vigilancia integral del paciente, ya que en caso de ET puede desarrollar otras complicaciones clínicas, por lo que consideramos necesaria la valoración por las diferentes especialidades incluyendo: genética, dermatología, neurología, oftalmología, cardiología, odontología, nefrología.

