











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkA 74-year-old woman without previous medical history except for the left bundle branch block was admitted for evaluation of recurrent syncopes. During admission, she experienced a sustained self-limited monomorphic ventricular tachycardia together with new syncope. Initial echocardiography displayed moderate biventricular systolic dysfunction. Cardiac magnetic resonance imaging (CMRI) confirmed these findings (Fig. 1) and revealed patchy subepicardial areas of late gadolinium enhancement within the left ventricular inferolateral and apical segments (red arrow) and an aneurysm was found in the right ventricular apex, containing a rounded thrombus (blue arrow) which persisted 10 days after intravenous anticoagulation therapy.

Figure 1 CMRI. Short axis. Inversion-recovery sequence. Late gadolinium enhancement in inferolateral wall (red arrow). Aneurysm in the right ventricle containing a rounded thrombus (blue arrow).
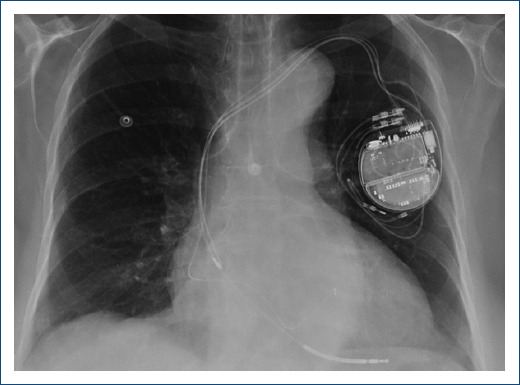
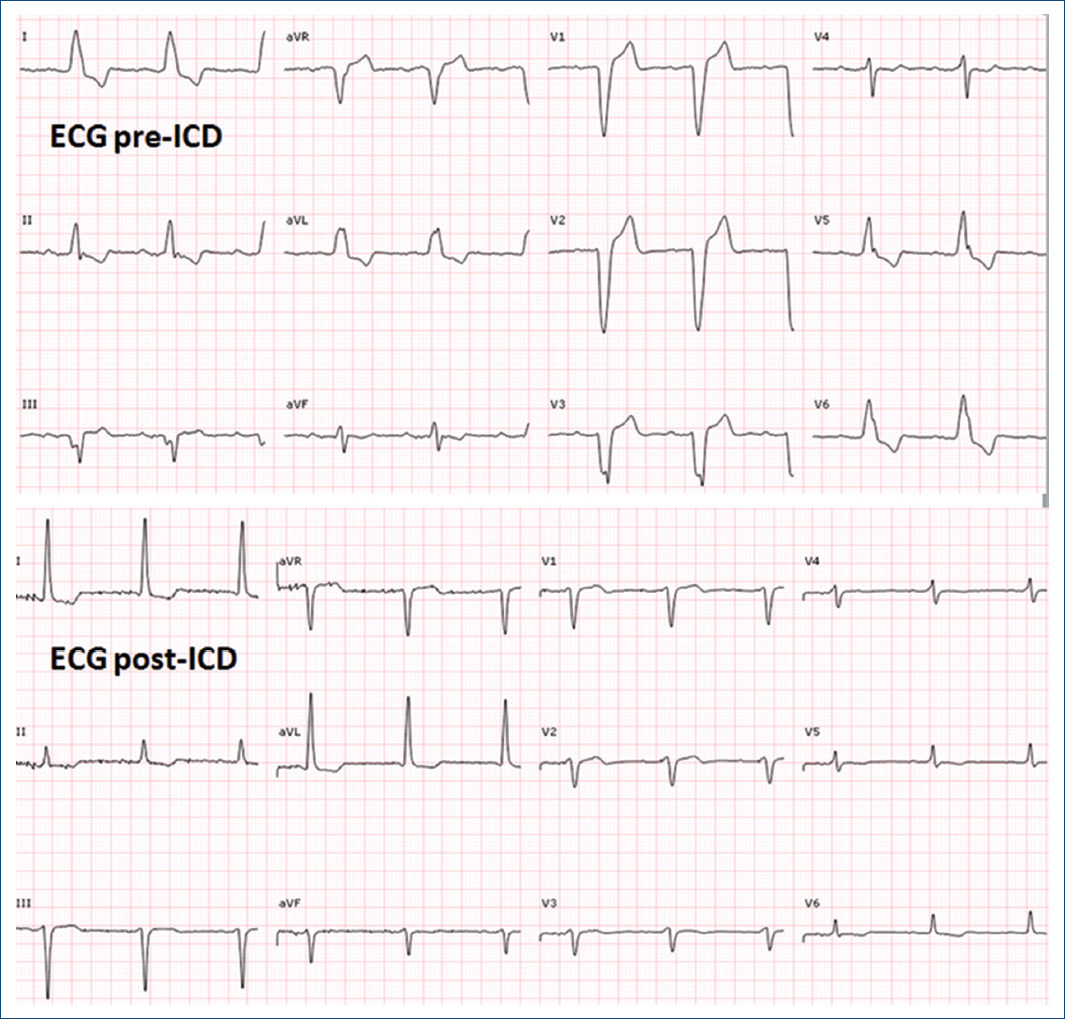
Due to the clinical suspicion of biventricular arrhythmogenic cardiomyopathy with sustained ventricular arrhythmias, cardioverter-defibrillator (ICD) implantation was decided. Subcutaneous approach, initially preferred due to persistent thrombus, was finally dismissed due to predicted high risk of inappropriate therapies in the screening test. Finally, a transvenous ICD was implanted with defibrillation electrode located in the posterior interventricular vein and left bundle branch pacing (Fig. 2) with a significant reduction of paced QRS duration (Fig. 3) and partial recovery of biventricular function during the follow-up. Subsequently, a genetic study was performed confirming a pathogenic variant in the DSG2 gene, which not only explained the phenotype but also allowed familiar cascade screening (Fig. 4).

Arrhythmogenic cardiomyopathy is characterized by replacement of myocardium by fibro-fatty tissue. It is associated with mutations in the genes that encode cardiac desmosomes, crucial proteins for the cardiomyocytes electromechanical connection1,2. Although usually diagnosed in youngsters, the greater availability of genetic tests and CMRI help to detect the late phenotypes, with crucial implications for the patient’s relatives.

