










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.62 no.5 México sep./oct. 2005
Caso clínico
Exostosis múltiple hereditaria y síndrome de Down
Multiple hereditary exostoses and Down's syndrome
Dr. Luis Gómez–Valencia1, Q.C. Anastasia Morales–Hernández2, Q.F.B. Josefina Salomón–Cruz2, Dr. Alfonso Jesús Berttolini–Díaz3, Dr. Ramón Miguel Cornelio–García4, Dr. Ezequiel Toledo–Ocampo5
1 División Académica de Ciencias de la Salud, Universidad Juárez Autónoma de Tabasco;
2 Laboratorio de Citogenética,
3 Servicio de Ortopedia y Traumatología,
4 Servicio de Anestesiología,
5 Servicio de Epidemiología, Hospital del Niño Dr. Rodolfo Nieto Padrón, Villahermosa, Tabasco, México.
Solicitud de sobretiros:
Dr. Luis Gómez Valencia,
Servicio de Genética del Hospital del Niño Dr. Rodolfo Nieto Padrón
Avenida Gregorio Méndez Magaña No. 2832, Col.Tamulté,Villahermosa,
Tabasco, México.
Fecha de recepción: 24–05–2005.
Fecha de aprobación: 13–10–2005.
Resumen
Introducción. La exostosis múltiple hereditaria es un trastorno autosómico dominante caracterizada por excrecencias cartilaginosas múltiples, fundamentalmente, en huesos de las extremidades, y en la que se han descrito como asociados el síndrome de Langer Giedion, la leucemia mieloide aguda y la espondilitis anquilosante. Objetivo: describir el caso de un niño de 10 años de edad en el cual coexisten la exostosis múltiple hereditaria y síndrome de Down.
Caso clínico. Paciente masculino de 10 años, con edad aparente mayor a la real, braquicefalia, fisuras palpebrales oblicuas, epicanto, puente nasal aplanado, retardo mental, con presencia de tumoraciones de 3 x 2 cm aproximados, localizadas en extremidades, cintura escapular y pélvica, con marcha claudicante y complemento cromosómico de 47, XY, +21.
Conclusión. Parece tratarse del primer caso en donde coexisten el síndrome de Down y la exostosis múltiple.
Palabras clave. Exostosis cartilaginosa múltiple; exostosis múltiple hereditaria; osteocondromatosis; síndrome de Down.
Abstract
Introduction. Multiple hereditary exostoses is an autosomal dominant disorder characterized by multiple osteochondromas, fundamentally in bones of the extremities, and in which they have been described like the associates the syndrome of Langer Giedion, the acute myeloid leukemia and the ankylosing spondylitis. Objective: to describe the case of 10–year–old boy in which coexist multiple hereditary exostoses and Down's syndrome.
Case report. Male patient with greater apparent age to the real one, brachycephaly, up slanting palpebral fissures, low nasal bridge, mental deficiency, and tumors presence of approximate 3 x 2 cm, located in extremities waist scapular and pelvic, with failing march and complement chromosomal of 47, XY, + 21.
Conclusion. We report appears to be the first case of Down's syndrome with the coexistence of multiple exostoses.
Key words. Exostoses, multiple hereditary; osteochondromatosis; Down's syndrome.
Introducción
La exostosis múltiple es un desorden caracterizado por excrecencias cartilaginosas múltiples, fundamentalmente de las diáfisis de huesos de las extremidades, pero que también pueden involucrar a huesos de la cintura escapular, pélvica y costillas, y en menor grado a vértebras, esternón, columna y huesos de carpo y tarso.1 Asociado a la exostosis múltiple se han descrito la metacondromatosis y el síndrome de Langer Giedion,2 la leucemia mieloide aguda3 y la espondilitis anquilosante.4 De acuerdo a la revisión de la literatura éste parece ser el primer caso reportado en que coexisten la exostosis cartilaginosa múltiple y el síndrome de Down.
Este trabajo tiene como objetivo presentar el caso de un varón de 10 años de edad con síndrome de Down y en el cual coexiste la exostosis múltiple familiar.
Presentación del caso clínico
Paciente masculino de 10 años de edad, originario y residente de Coatzacoalcos, Veracruz, México, producto de la tercera gesta, embarazo normoevolutivo, de término, parto aparentemente normal, atendido en su domicilio por empírica. Período neonatal sin complicaciones, desconociéndose somatometría al nacimiento. El desarrollo neurológico en su infancia fue retardado: sostén cefálico al año de vida, sedestación a los 18 meses, deambulación a los dos años, inició lenguaje a los tres años de edad sin lograr hasta el momento pronunciación de frases.Ambos padres de 31 años de edad al momento de nacer el propositus, no consanguíneos y con antecedentes de exostosis múltiple en la abuela paterna (I–2), el padre (II–2), dos tíos paternos (II–5,6), tres hermanos (III–1,2 y 4), y en ocho primos hermanos (III–5–10, 12–13) (Fig. 1).
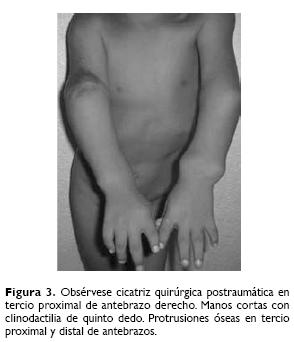
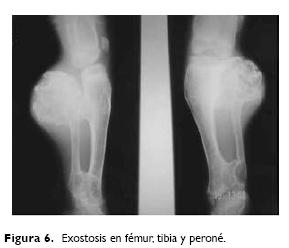
A la exploración física: peso 39 kg, talla 117 cm, edad aparente mayor a la real, cráneo braquicéfalo, fisuras palpebrales oblicuas, epicanto, puente nasal aplanado, implantación baja de pabellones auriculares, micrognatia, cuello corto, manos cortas, con equivalente de pliegue transverso palmar, clinodactilia de quinto dedo, acortamientos de extremidades y talla baja, con deformaciones en regiones proximales y distales, con limitación a los movimientos de flexión y extensión (Fig. 2). En extremidad superior derecha presenta cicatriz postquirúrgica en tercio proximal y lateral del antebrazo, tumoraciones en tercio proximal y distal de ambos brazos y antebrazos, así como en tercio proximal y distal de muslos y piernas de aproximadamente 3 x 2 cm, incapacitantes y dolorosos, miembro pélvico izquierdo en valgo (Figs. 3 y 4). Los estudios de rutina consistentes en: biometría hemática, química sanguínea y examen general de orina resultaron normales. El cultivo de linfocitos de sangre periférica para análisis cromosómico mostró un complemento 47, XY, +21, no habiéndose reportado aberraciones de tipo estructurales. Radiológicamente se observó exostosis cartilaginosas múltiples metafisarias de huesos largos, cortos y planos, de formas y tamaños variables, desde las pequeñas exostosis longilíneas hasta las exostosis vegetativas voluminosas que deforman las articulaciones (Figs. 5 y 6).



Discusión
La exostosis múltiple hereditaria es un desorden esquelético caracterizado por múltiples osteocondromas que desencadenan dolor, deforman la región y potencializan la degeneración maligna.1 El moldeamiento de los huesos es anormal, particularmente los huesos largos, manifestándose con acortamiento de talla e irregularidades de corticales y metáfisis. La compresión e irritación de nervios adyacentes, vasos y tendones, así como la obstrucción urinaria e intestinal son complicaciones que suelen presentarse. Además de las diáfisis, también pueden estar involucrados los huesos de la cintura escapular, pélvica y costillas, y en menor grado las vértebras, esternón, columna y huesos de carpo y tarso.2,3 Vanhoenacker y col.3 estudiaron los aspectos clínicos y radiológicos en 31 afectados con exostosis múltiple, correspondientes a dos familias en cuatro generaciones,y encontraron que los sitios comunes de localización de exostosis fueron la región frontal y craneal, mandíbula, húmero, columna, cadera, rodillas, tibia y peroné. En el caso que nos ocupa los sitios de afección en el propositus es a nivel de cadera, rodillas, tibia y peroné; sin embargo, la revisión clínica en dos de sus hermanas (III–1–2) mostró sitios no comunes de afección como falanges de manos en su tercio proximal y omóplato derecho.
Desde el punto de vista hereditario la exostosis múltiple es un trastorno autosómico dominante, con una prevalencia estimada en 1/50 000, y con un porcentaje de malignización de las lesiones de aproximadamente 2%,4 y en la cual están implicados tres tipos de genes: EXT 1, EXT 2 y el EXT 3. Al gen EXT 1 se le conoce como gen tumor supresor; cerca de 70% de los casos de exostosis múltiple presentan mutación del gen EXT 1, localizado en el cromosoma 8 (8q24), mientras que 30% de las familias presentan mutación del gen EXT 2 localizado en el cromosoma 11 (11 p1 1–p12). El gen EXT 3 se encuentra localizado en el cromosoma 19 (19p).4,5
Francannet y col.6 en un estudio de 42 pacientes con exostosis múltiple hallaron que en 90% de los casos los genes EXT 1 y EXT 2 codifican para la glucosiltransferasa involucrada en la biosíntesis del heparán sulfato. Estos autores encontraron relación de asociación del EXT 1 con la gravedad de la enfermedad y su transformación maligna, sugiriendo que la correlación genotipo–fenotipo facilita el manejo de estos pacientes.
Asociado a la exostosis múltiple se han descrito la metacondromatosis y el síndrome de Langer Giedion,2 la leucemia mieloide aguda3,7,8 y la espondilitis anquilosante.4,9 La única comunicación que involucra indirectamente al síndrome de Down es la de Niikawa,8 la cual señala que existen evidencias de genes responsables tanto para la mielopoyesis anormal, como para la exostosis cartilaginosa múltiple. La mielopoyesis anormal es una reacción leucemoide que ocurre frecuentemente en niños con síndrome de Down, los cuales a menudo desarrollan leucemia grave años más tarde. El autor enfatiza que tanto los genes de la mielopoyesis anormal como los de la exostosis cartilaginosa múltiple están involucrados en anomalías cromosómicas de tipo estructural como la translocación de novo 8q–13q, y la invasión en el cromosoma 21 (q11.1q22.13). En el presente trabajo el cariotipo realizado con técnica convencional no mostró aberraciones de tipo estructural.
Olmez y col.9 mencionan que existen también padecimientos con lesiones parecidas a la exostosis como son el síndrome de Ehlers–Danlos y la hipofosfatemia hereditaria, pero que en estos casos las exostosis están localizadas en sitios de músculos y tendones. Los mismos autores, al reportar el caso de un varón de 50 años de edad con exostosis múltiple hereditaria y espondilitis anquilosante, consideran que para los diferentes mecanismos genéticos no es posible diferenciar entre coincidencia y asociación, la coincidencia presupone un acto fortuito y la asociación involucra mecanismos fisiopatológicos inherentes; la coexistencia es un término que no descarta categóricamente una asociación, sin embargo, los autores sugieren que la coexistencia representa más una coincidencia que una asociación. Otros diagnósticos diferenciales son: anetodermia y metacondromatosis, y la encondromatosis múltiple o enfermedad de Ollier.
El caso aquí informado corresponde a un niño con síndrome de Down, y el cual presenta exostosis múltiple en regiones corporales frecuentes, de mayor gravedad que en los otros miembros de la familia con padecimiento similar. Se coincide con Olmez y col.9 en que este evento pudiera representar más una coincidencia que una asociación. Después de realizar una búsqueda en revistas científicas indexadas, los autores del presente trabajo no encontraron ningún reporte de coincidencia de síndrome de Down y exostosis múltiple hereditaria, por lo que se supone representa el primer caso descrito relacionado con estas dos entidades.10–14
Referencias
1. Pierz KA, Stieber R, Kusumi K, Dormans JP. Hereditary multiple exostoses: One center's experience and review of etiology. Clin Orthop. 2002; 401:49–59. [ Links ]
2. Mckusick VA. Mendelian inheritance in man. A catalog of human genes and genetic disorders. 12da ed. New York: The Johns Hopkins University Press; 1988.Vol. 2. p. 634–63. [ Links ]
3. Vanhoenacker FM, van Hul W, Wuyts W. Hereditary multiple exostoses: from genetics to clinical syndrome and complications. Eur J Radiol. 2001; 40:208–17. [ Links ]
4. Rambeloarisoa J, Guedy M, Legeai ML. Hereditary multiple exostoses after 40 years of development: a case report. Rev Med Interne. 2002; 23: 657–64. [ Links ]
5. Wuyts W, Bovee JU, Hogendoorn PC. From gene to disease; hereditary multiple exostoses. Ned Tijdschr Geneeskd. 2002; 145: 162–4. [ Links ]
6. Francannet C, Cohen–Tanugi A, Le Merrer M. Genotype–phenotype correlation in hereditary multiple exostoses. J Med Genet. 2001; 38:430–4. [ Links ]
7. Gozdasoglu S, Vysal Z, Kurekci, AE. Hereditary multiple exostoses and acute myeloid leukemia: an unusual association. Pediatr Hematol Oncol. 2000; 17: 707–11. [ Links ]
8. Niikawa N. Positional cloning of the putative gene responsible for transient and myelopoiesis and that for multiple cartilaginous exostoses. Rinsho Byori. 1996; 44: 13–8. [ Links ]
9. Olmez N, Gunaydin R, Gurgan A, Elcin F. Coexistence of hereditary multiple exostoses and ankylosing spondylitis. Clin Rheumatol. 1999; 18:481–4. [ Links ]
10. Bamba I, Sie–Essoh JB, Kacou DA. Multiple exostoses. Review of the literature. Apropos of a case disclosed. Bull Soc Pathol Exot. 2002; 95: 83–5. [ Links ]
11. Toguehida J, Nagayama S. Multiple exostoses. Nippon Rinsho. 2000; 58: 1473–8. [ Links ]
12. Wicklund CL, Pauli RM, Johnstan–Dhecht JT. Natural history study of multiple hereditary exostoses. Am J Med Genet. 1995; 55:43–6. [ Links ]
13. Polchau H, Johannson W, Suttorp M, Rister M. Isolated trisomy 21 in blasts of an acute lymphoblastic leukemia. Monatsschr Kinderheilkd. 1987; 135:220–2. [ Links ]
14. Arpino C, Piciullo A, Palmarino M. Lack of association between IDE genetic variability and Down's syndrome. Neurosci Lett. 2005; 382: 93–5. [ Links ]

