










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.70 no.1 México ene./feb. 2013
ARTÍCULOS DE INVESTIGACIÓN
Prevalencia de mosaicismo para la trisomía 21 y análisis de las variantes citogenéticas en pacientes con diagnóstico de síndrome de Down. Revisión de 24 años (1986-2010) del Servicio de Genética del Hospital General de México ''Dr. Eduardo Liceaga''
Prevalence of mosaicism for trisomy 21 and cytogenetic variant analysis in patients with clinical diagnosis of Down syndrome: a 24-year review (1986-2010) at the Servicio de Genética, Hospital General de México ''Dr. Eduardo Liceaga''
Luz María Garduño-Zarazúa,1 Lucila Giammatteo Alois,2 Susana Kofman-Epstein,1,3 Alicia B. Cervantes Peredo1,3
1 Servicio de Genética, Hospital General de México ''Dr. Eduardo Liceaga''
2 Facultad de Estudios Superiores, Cuautitlán
3 Facultad de Medicina. Universidad Nacional Autónoma de México
México, D.F., México
Autor de correspondencia: M. en C. Alicia B. Cervantes Peredo
Correo electrónico: acervant@unam.mx
Fecha de recepción: 03-07-12
Fecha de aceptación: 21-01-13
Resumen
Introducción. El síndrome de Down es la causa genética más frecuente de discapacidad intelectual, con incidencia de 1/650 recién nacidos. La determinación de la variante citogenética es importante para el asesoramiento. En este trabajo se determinó la prevalencia de las variantes citogenéticas en los pacientes con síndrome de Down del Hospital General de México y se analizaron con relación a la edad materna.
Métodos. Se revisaron los resultados de los cariotipos realizados por indicación de síndrome de Down de enero 1986 a diciembre 2010. Se obtuvieron las edades de los pacientes y de sus madres.
Resultados. De 581 pacientes, 71 (12.22%) presentaron cariotipo normal. De los 510 confirmados para síndrome de Down, 445 (87.3%) resultaron con trisomía regular; 22 (6.3%), como producto de una translocación robertsoniana; y 43 (8.4%), mosaico. La edad materna en aquellos con trisomía regular (mediana de 30 años) y mosaicismo (mediana de 29 años) fue mayor que en los debidos a una translocación (mediana de 20 años).
Conclusiones. Caracterizar la aberración cromosómica en pacientes mexicanos con síndrome de Down permitió dar un asesoramiento genético adecuado y establecer la prevalencia de mosaicismo en nuestra población, que resultó mayor a lo reportado en la literatura. Es importante detectar mosaicos en pacientes cuyos datos clínicos no confirmen síndrome de Down o presenten discapacidad intelectual de causa desconocida.
Palabras clave: síndrome de Down, trisomía 21, mosaicismo, aberración cromosómica.
Abstract
Background. Down syndrome is the principal genetic cause of learning disabilities, with an incidence of 1/650 live births. Diagnosis is confirmed by karyotyping. Chromosomal variants are important for genetic counseling. We determined the prevalence of cytogenetic variants in patients with clinically diagnosed Down syndrome in the Hospital General de México Dr. Eduardo Liceaga and discussed its relevance according to maternal age.
Methods. We reviewed karyotype data of patients with clinically diagnosed Down syndrome from January 1986 to December 2010 and obtained maternal and patient ages.
Results. From 581 patients analyzed, 71 (12.22%) had normal karyotype. In 510 patients we confirmed that 445 (87.3%) had a regular trisomy, 22 (6.3%) were the product of a Robertsonian translocation and mosaicism was found in 43 (8.4%) patients. Maternal age was higher in patients with regular trisomy (median: 30 years) and mosaicism (median: 29 years) than in those with translocations (median: 20 years).
Conclusions. Characterization of chromosomal aberrations in Mexican DS patients allows us to offer appropriate genetic counseling and to establish the prevalence of mosaicism in our population, which was higher than the reported data. Cytogenetic analysis for detection of mosaicism is important in patients with scarce clinical data of DS or with learning disabilities of unknown origin.
Key words: Down syndrome, trisomy 21, mosaicism, chromosomal aberrations.
Introducción
El síndrome de Down (SD) es la causa genética más frecuente de discapacidad intelectual en la población y se debe a un efecto de dosis génica por la presencia de un cromosoma 21 adicional1 o de una trisomía parcial, principalmente de la región 21q22.2 Los pacientes presentan un fenotipo característico que incluye braquicefalia, facies plana, fisuras palpebrales oblicuas ascendentes, epicanto, pabellones auriculares redondos con pliegues anormales e implantación baja, hipotonía y pliegue palmar transverso único, entre otros.3 También pueden presentar enfermedades cardiacas (40-50%) y atresia duodenal, además de un incrementado riesgo de leucemia mieloide aguda, enfermedad de Hirschprung y de enfermedad de Alzheimer (especialmente después de la cuarta década).1
En México, el SD presenta una incidencia de 1/650 recién nacidos vivos.3 El diagnóstico se realiza con base en las características clínicas. Sin embargo, es recomendable confirmarlo por cariotipo, para determinar la variante citogenética y dar un adecuado asesoramiento genético. El SD presenta cinco variantes citogenéticas:
• Trisomía 21 regular (T21). Con cariotipo 47,XX,+21 o 47,XY,+21, presente en alrededor de 95% de los casos.2,4
• Translocaciones robertsonianas (rob). Involucran el rearreglo de un cromosoma 21 con otro de los cromosomas acrocéntricos (de los grupos D o G).2,4 Su fórmula es 46,XX o XY,rob(D o G;21)(q10;q10),+21.
• Isocromosomas del brazo largo del cromosoma 21. Con cariotipo 46,XX o XY,+21,i(21)(q10). Junto con las rob, se presentan en aproximadamente 4% de los pacientes con SD.2,4
• Mosaicismo. Se define como la presencia de dos o más líneas celulares diferentes en el mismo individuo. En este caso, una línea con T21 y otra línea normal. Se representa con la fórmula 47,XX o XY,+21/46,XX o XY y corresponde a 1-3% de todos los casos.2,4
• Trisomía parcial de la región 21q22.3. Con cariotipo 46,XX o XY,dup(21)(q22.3). Se observa en menos de 1% de los casos.2,4
La T21 es frecuentemente resultado de una no disyunción en la meiosis materna (~90%). La mayoría ocurre en meiosis I (MI). Los errores de meiosis II (MII) solamente constituyen 20% de los errores maternos.2 Las trisomías de origen paterno son menos frecuentes (3% en MI y 5% en MII)5 y, en alrededor de 4% de los casos, el cromosoma adicional es el resultado de un error postcigótico.6 Es importante mencionar que el porcentaje de mosaicos puede estar subdiagnosticado, dado que el número de células analizadas generalmente es insuficiente para detectar líneas celulares en baja proporción. Los mosaicos en el SD pueden originarse de un cigoto normal o a partir de uno trisómico por la no disyunción postcigótica o por rescate trisómico, respectivamente.7
Este estudio se realizó con el objetivo de analizar, en forma retrospectiva, los datos citogenéticos de los pacientes del Hospital General de México Dr. Eduardo Liceaga (HGM) con sospecha diagnóstica de SD y/o cariotipo confirmado, determinar la prevalencia de las variantes citogenéticas en esta población con SD y analizarlas en relación con la edad materna. Cabe señalar que existen pocos reportes de la prevalencia de mosaicismo en SD en la población mexicana.
Métodos
Se recopilaron los datos de los pacientes con diagnóstico de SD que acudieron al Servicio de Genética del Hospital General de México (HGM) durante el período de 1986 a 2010. Se registraron el resultado citogenético, el sexo, la edad y los datos clínicos del propósito y la edad materna al nacimiento del mismo. Se calcularon las medidas de tendencia central para las edades de los pacientes y las maternas. Para el cariotipo, las muestras fueron procesadas y bandeadas por las técnicas citogenéticas convencionales. Se analizaron de 20 a 100 metafases por caso, dependiendo de la indicación y de la calidad del material.
Resultados
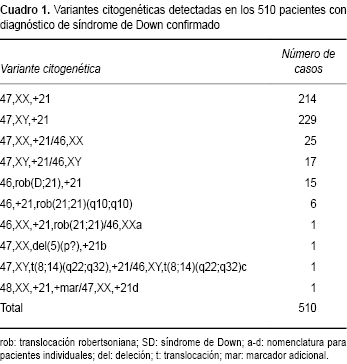
Durante los 24 años de estudio, se registraron 581 pacientes con indicación diagnóstica de SD y resultado de cariotipo; 71 pacientes presentaron cariotipo normal (Cuadro 1). La alteración cromosómica predominante fue la T21, seguida de mosaicos y rob (Figura 1). En la mayoría de los mosaicos la línea celular más abundante fue la trisómica. La proporción promedio de la línea anormal fue 72%, con un rango de 2 a 98%. Sólo en dos pacientes, con discapacidad intelectual y pocos datos clínicos de SD, la línea trisómica correspondió a menos de 10% (2 y 8%). En pocos casos debidos a translocación fue posible estudiar a los padres, y en su mayoría correspondieron a rearreglos de novo . Con respecto a la edad de los pacientes al momento de la consulta, no se observó una variación significativa (Cuadro 2). En cuanto a la edad materna, fue mayor en los pacientes con T21 y mosaicismo que en los pacientes por rob (Figura 2).


Discusión
Una no disyunción meiótica materna ocurre en ~90% de los casos de SD, lo cual se correlaciona con que la variante citogenética más frecuente sea la T21 (445 pacientes) (Figura 1). Otro mecanismo para esta aneuploidía es que alguno de los padres, principalmente la madre, sea mosaico. Se estima que 3% de las parejas que tienen un hijo con T21 presentan mosaicismo,8 y que 1-2% de la población general adulta presenta mosaicismo para autosomas.9 En México, se ha reportado que 2.36% de los progenitores de hijos con SD son mosaicos con una línea con T21 en muy baja proporción (2.7-4.3%).10 En estudios recientes se ha detectado mosaicismo en 4.5% de los padres con un hijo con T21, y hasta en 36% de los padres con recurrencia de SD.2 Hultén y colaboradores encontraron en fetos femeninos, aparentemente normales, mosaicos germinales para trisomía 21; esto sugiere que el efecto de la edad materna es causado por la selección diferencial de estas células durante el desarrollo fetal y postnatal hasta la ovulación. Un alto grado de mosaicismo germinal puede explicar la existencia de mujeres jóvenes que tienen recurrencia de hijos con SD.4 El riesgo de recurrencia para T21 en una familia con un hijo afectado es de 1-2%, y puede incrementarse por la edad materna.3
Con respecto a las rob (Cuadro 1, Figura 1), la proporción encontrada fue la esperada de acuerdo con la literatura. Sin embargo, no fue posible establecer la prevalencia de cada tipo de translocación. Algunos estudios epidemiológicos indican que la translocación más frecuente en SD es la rob(14;21), seguida por la rob(21;21) o i(21q).11,12 Cuando un paciente con SD presenta alguna de estas variantes es necesario realizarles un cariotipo a los padres, para identificar si uno de ellos es portador, y establecer un riesgo de recurrencia adecuado. La mayoría de las traslocaciones ocurren de novo . La frecuencia reportada de portadores es de 5 a 20% y, generalmente, la madre es la portadora.12-14 En nuestro estudio, aun cuando en pocos casos fue posible estudiar citogenéticamente a los padres, los resultados concuerdan con lo antes señalado. El riesgo de recurrencia es menor a 1% si la translocación es de novo. 12 En caso de ser heredada, el riesgo empírico para las rob(D;21) cuando la madre es portadora es de ~10-20% y cuando es el padre, de ~2-5%. Para un portador de una rob(21;21), el riesgo de recurrencia es de 100%.15
La prevalencia de mosaicismo en este estudio fue más elevada que la reportada en la literatura,2-4 pero coincide con otros estudios epidemiológicos en pacientes con SD (Cuadro 3).7,12-14,16-22 Los pacientes con SD en mosaico con una línea celular trisómica en proporción mayor a 90% suelen diagnosticarse como trisomía regular ya que, a menos que se analicen >50 células, no se detecta la línea normal. Por el contrario, si la línea trisómica se encuentra en proporción menor a 10%, el diagnóstico con frecuencia pasa inadvertido.7,23 Al analizar 25 metafases, se detectan líneas en proporciones >25%.23 De esta manera, se considera que la frecuencia de mosaicismo en SD reportada (1-3%) es menor a la real.7 En este trabajo se cuantificaron, en promedio, 38 células por caso, con un intervalo de 20-100 células.
La mayoría de los mosaicos encontrados en este (Cuadro 1) y otros estudios (Cuadro 3) presentan solo una línea trisómica y otra normal. Aunque no se conocen las proporciones, es de esperarse que exista predominio de la línea trisómica. González y colaboradores estudiaron 100 pacientes mexicanos con SD. Analizaron 100 metafases y encontraron mosaicismo en 39 de ellos, 13 con 97% de línea trisómica, en 35 la línea con T21 en proporción mayor de 90%, y sólo en 4 menor de 50%.7 En nuestro estudio, sólo dos pacientes de 43 en mosaico tuvieron la línea trisómica en proporción menor de 10% y presentaron discapacidad intelectual con algunos rasgos de SD. En los demás, la proporción de la línea celular con T21 fue igual a la de la normal (50%) o superior. Se ha establecido que varios individuos diagnosticados como SD típico en realidad son mosaicos con una pequeña proporción de células normales. En linfocitos cultivados; también se ha demostrado que algunos individuos con rasgos mínimos de SD, e incluso sin ellos, pueden ser mosaicos en bajo grado con variaciones en las proporciones en otros tejidos.19,24
Cuando se sospecha o detecta mosaicismo, se recomienda buscar la línea trisómica en, por lo menos, dos tejidos. Se ha observado que el número de células anormales en la mucosa oral está relacionado significativamente con el coeficiente intelectual (células derivadas del ectodermo). En cambio, los defectos cardiacos se correlacionan con la proporción encontrada en linfocitos, ya que ambos tejidos derivan del mesodermo.25 Dado que este estudio fue retrospectivo, no se verificó la presencia de mosaicismo en otros tejidos, aunque se considera realizarlo en estudios posteriores.
De las variantes atípicas identificadas (Cuadro 1, pacientes a-d), el primer paciente (a) presentó una rob(21;21) o i(21q) en mosaico con una línea normal. Se infiere que se debe a un error postcigótico; sin embargo, es necesario realizar pruebas moleculares y citogenéticas para comprobarlo, descartar quimerismo y establecer el origen de la aberración estructural. El paciente (b), además de la T21, presentaba monosomía parcial de 5p, por lo que su fenotipo estaba modificado con rasgos clínicos del síndrome de Cri du Chat (maullido de gato). El paciente (c), con una translocación recíproca balanceada t(8;14)(q32;q22) constitutiva, fue mosaico para la T21. El rearreglo aparentemente balanceado no repercutió en el fenotipo; no obstante, es importante realizar el cariotipo a los padres para descartar que alguno sea portador. El último paciente (d), además de la T21, presentó un cromosoma marcador adicional en mosaico del cual debe realizarse la caracterización citogenética, ya que su presencia puede modificar el fenotipo y repercutir en el asesoramiento genético. En otros estudios, también se han reportado alteraciones cromosómicas adicionales a la T21.11,13
La edad de los pacientes en el momento del diagnóstico presentó un rango desde recién nacido (RN) a 52 años y una mediana de 3 meses, y no se observó diferencia significativa. La mayoría de los pacientes son referidos a la consulta de Genética por diferentes especialistas, principalmente neonatólogos y pediatras, debido a su fenotipo. En las edades maternas, se observó una diferencia significativa entre las variantes citogenéticas. La edad materna de las T21 (mediana 30 años) y mosaicos (mediana 29 años) fue mayor a la de las rob (mediana 20 años) (Cuadro 2). En la literatura se reporta un comportamiento similar, siendo más de 35 años para T21 y menos de 25 para rob. La edad materna reportada para la T21 es mayor que la observada en nuestros datos, lo cual podría explicarse por las conductas reproductivas.11,14,16,18,26 El SD por rob se debe generalmente a una anomalía estructural de novo o la segregación de esta alteración en portadores sanos, y no a errores en la disyunción cromosómica, por lo que no está asociada a un efecto de edad materna avanzada.11,14
En la Figura 2 se observa que el número de casos por edad materna avanzada fue mayor en T21 que en las otras alteraciones. Se presentan dos picos en las T21 y los mosaicos: el primero corresponde a la edad promedio de reproducción y el segundo indica el aumento de casos por efecto de la edad materna, lo que concuerda con la literatura.11,16,17 Dado que los mosaicos pueden originarse por la no disyunción postcigótica o por rescate trisómico, también tienen un efecto de edad materna, por lo que su comportamiento es similar a la T21.3,5 En el caso de las rob sólo existe un pico que corresponde a la edad reproductiva promedio. En cuanto a los pacientes con fenotipo de SD y cariotipo normal, sería conveniente realizar estudios moleculares como FISH (de sus siglas en inglés, Fluorescent In Situ Hybridization ) con sonda para la región 21q22 en diferentes tejidos, para descartar mosaicismo en baja proporción o rearreglos crípticos.
En este trabajo, la revisión de los datos citogenéticos permitió caracterizar la prevalencia de los diferentes tipos de alteraciones citogenéticas en SD en nuestra población. Este reporte es, al día de hoy, el que incluye el mayor número de pacientes mexicanos estudiados. Los datos obtenidos resultan de suma importancia para calcular los riesgos de recurrencia y dar un asesoramiento genético adecuado. Dado que la prevalencia de mosaicismo fue mayor que la reportada en la literatura, cabe recalcar la importancia de realizar una búsqueda adecuada en individuos con pocos rasgos de SD o con discapacidad intelectual de origen desconocido.
REFERENCIAS
1. Vundinti BR, Ghosh K. Incidence of Down syndrome: hypotheses and reality. Indian J Hum Genet 2011;17:117-119. [ Links ]
2. Frias S, Ramos S, Molina B, del Castillo V, Mayén DG. Detection of mosaicism in lymphocytes of parents of free trisomy 21 offspring. Mutat Res 2002;520:25-37. [ Links ]
3. Secretaría de Salud. Centro Nacional de Equidad de Género y Salud Reproductiva. Atención Integral de la Persona con síndrome de Down. Lineamiento Técnico. Secretaría de Salud 2007. Disponible en: http://www.salud.gob.mx/unidades/cdi/documentos/Sindrome_Down_lin_2007.pdf [ Links ]
4. Hultén MA, Patel SD, Tankimanova M, Westgren M, Papadogiannakis N, Jonsson AM, et al. On the origin of trisomy 21 Down syndrome. Mol Cytogenet 2008;1:21. doi: 10.1186/1755-8166-1-21. [ Links ]
5. Vekemans M. Trisomy. Encyclopedia of Life Sciences (ELS). Chichester: John Wiley & Sons, Ltd; 2005. doi: 10.1038/npg.els.0005544. [ Links ]
6. Girirajan S. Parental-age effects in Down syndrome. J Genet 2009;88:1-7. [ Links ]
7. González-Herrera L, Pinto-Escalante D, Ceballos-Quintal JM. Prevalencia de mosaicos en 100 individuos con diagnóstico de síndrome de Down. Rev Biomed 1998;9:214-222. [ Links ]
8. Harris DJ, Begleiter ML, Chamberlin J, Hankins L, Magenis RE. Parental trisomy 21 mosaicism. Am J Hum Genet 1982;34:125-133. [ Links ]
9. Iourov IY, Vorsanova SG, Yurov YB. Chromosomal mosaicism goes global. Mol Cytogenet 2008;1:26. doi: 10.1186/1755-8166-1-26. [ Links ]
10. Armendares S, Buentello L, Salamanca F. Cytogenetic study of the parents of 85 index cases with regular trisomy 21. Rev Invest Clin 1990;42:180-188. [ Links ]
11. Mutton D, Alberman E, Hook EB. Cytogenetic and epidemiological findings in Down syndrome, England and Wales 1989 to 1993. National Down Syndrome Cytogenetic Register and the Association of Clinical Cytogeneticists. J Med Genet 1996;33:387-394. [ Links ]
12. Sheth F, Rao S, Desai M, Vin J, Sheth J. Cytogenetic analysis of Down syndrome in Gujarat. Indian Pediatr 2007;44:774-777. [ Links ]
13. Thomas IM, Rajangam S, Hegde S. Cytogenetic investigations in Down syndrome patients and their parents. Indian J Med Res 1992;96:366-371. [ Links ]
14. Chandra N, Cyril C, Lakshminarayana P, Nallasivam P, Ramesh A, Gopinath PM, et al. Cytogenetic evaluation of Down syndrome: a review of 1020 referral cases. Int J Hum Genet 2010;10:87-93. [ Links ]
15. Luthardt FW, Keitges E. Chromosomal syndromes and genetic disease. Encyclopedia of Life Sciences (ELS). Chichester: John Wiley & Sons, Ltd. 2001. [ Links ]
16. Astete C, Youlton R, Castillo S, Be C, Daher V. Análisis clínico y citogenético en 257 casos de síndrome de Down. Rev Chil Pediatr 1991;62:99-102. [ Links ]
17. Jyothy A, Kumar KS, Rao GN, Rao VB, Swarna M, Devi BU, et al. Cytogenetic studies of 1001 Down syndrome cases from Andhra Pradesh, India. Indian J Med Res 2000;111:133-137. [ Links ]
18. Staples AJ, Sutherland GR, Haan EA, Clisby S. Epidemiology of Down syndrome in South Australia, 1960-89. Am J Hum Genet 1991;49:1014-1024. [ Links ]
19. Devlin L, Morrison PJ. Accuracy of the clinical diagnosis of the Down syndrome. Ulster Med J 2004;73:4-12. [ Links ]
20. Azman BZ, Ankathil R, Siti Mariam I, Suhaida MA, Norhashimah M, Tarmizi AB, et al. Cytogenetic and clinical profile of Down syndrome in Northeast Malaysia. Singapore Med J 2007;48:550-554. [ Links ]
21. Catovic A, Kendic S. Cytogenetic findings at Down syndrome and their correlation with clinical findings. Bosn J Basic Med Sci 2005;5:61-67. [ Links ]
22. Wang YF, Lin L, Chen ZY. Cytogenetic study of Down syndrome cases in southern Hainan Province and report of a rare case of abnormal karyotype. Nan Fang Yi Ke Da Xue Xue Bao 2010;30:2592-2595. [ Links ]
23. Hook EB. Exclusion of chromosomal mosaicism: tables of 90%, 95% y 99% confidence limits and comments on use. Am J Hum Genet 1977;29:94-97. [ Links ]
24. Hultén MA, Jonasson J, Nordgren A, Iwarsson E. Germinal and somatic trisomy 21 mosaicism: how common is it, what are the implications for individual carriers and how does it come about? Curr Genomics 2010;11:409-419. [ Links ]
25. Papavassiliou P, York TP, Gursoy N, Hill G, Nicely LV, Sundaram U, et al. The phenotype of persons having mosaicism for trisomy 21/Down syndrome reflects the percentage of trisomic cells present in different tissues. Am J Med Genet A 2009;149A:573-583. doi: 10.1002/ajmg.a.32729. [ Links ]
26. Mokhtar MM, Abd el-Aziz AM, Nazmy NA, Mahrous HS. Cytogenetic profile of Down syndrome in Alexandria, Egypt. East Mediterr Health J 2003;9:37-44. [ Links ]

