










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.70 no.1 México ene./feb. 2013
CASO CLÍNICO
Atrofia muscular espinal tipo 1: enfermedad de Werdnig-Hoffmann
Type I spinal muscular atrophy: Werdnig-Hoffmann disease
Romeo Zárate-Aspiros,1 Ana Beatriz Rosas-Sumano,2 Alberto Paz-Pacheco,3 Patricia Fenton-Navarro,4 Silvet Chinas-López,4 José Antonio López-Ríos5
1 Servicio de Urgencias Pediatría, Hospital General de Oaxaca ''Dr. Aurelio Valdivieso''
2 Centro de Investigación Médica y Biológica, Facultad de Medicina y Cirugía, Universidad Autónoma Benito Juárez de Oaxaca
3 Departamento de Enseñanza, Hospital General de Oaxaca ''Dr. Aurelio Valdivieso''
4 Departamento de Genética, Hospital General de Oaxaca ''Dr. Aurelio Valdivieso''
5 Residente de Pediatría, Hospital General de Oaxaca ''Dr. Aurelio Valdivieso''
Oaxaca, Oaxaca, México
Autor de correspondencia: Dra. Ana Beatriz Rosas Sumano
Correo electrónico: arosumx@yahoo.com.mx
Fecha de recepción: 03-07-12
Fecha de aceptación: 08-11-12
Resumen
Introducción. Las atrofias musculares espinales de la infancia son enfermedades neuromusculares hereditarias, autosómicas, recesivas, caracterizadas por la degeneración de las neuronas motoras del asta anterior de la médula espinal. La atrofia muscular espinal tipo I (enfermedad de Werdnig-Hoffmann) es la forma más severa. Se inicia in útero o durante los primeros meses de vida. La muerte suele ocurrir antes de los dos años de edad.
Caso clínico. Lactante de 6 meses de edad que ingresa al Servicio de Urgencias por dificultad respiratoria severa. Presenta marcada hipotonía muscular, debilidad de musculatura intercostal y fasciculaciones de la lengua. La electromiografía es compatible con polineuropatía motora con daño mielínico y axonal. El análisis molecular reportó un estado homocigoto para la deleción de los exones 7 y 8 del gen SMN-1 . Con estos dos estudios se integra el diagnóstico de atrofia muscular espinal tipo 1 (enfermedad de Werdnig-Hoffmann).
Conclusiones. Es importante conocer y diagnosticar esta entidad para brindar consejo genético a la familia, así como asesoramiento y apoyo en el manejo del paciente.
Palabras clave: atrofia muscular espinal tipo I, enfermedad de Werdnig-Hoffmann.
Abstract
Background. Childhood spinal muscular atrophy is an autosomal recessive neuromuscular disease characterized by degeneration of the anterior horn cells of the spinal cord. SMA type I, the most severe form (Werdnig-Hoffmann disease) can be detected in utero or during the first months of life. Death typically occurs within the first 2 years of life.
Case report. A 6-month-old female was admitted to the emergency room for severe respiratory distress. She had muscular hypotonia, intercostal muscle weakness and tongue fasciculations. Electromyography was compatible with motor polyneuropathy with axonal and myelin damage. Molecular analysis of SMN-1 gene reported homozygous for deletion of exons 7 and 8 of SMN-1 gene.
Conclusions. It is imperative to recognize and diagnose this entity in order to provide genetic counseling to the family as well as to offer support and advice in the care of the patient.
Key words: type I spinal muscular atrophy, Werdnig-Hoffmann disease.
Introducción
Bajo la denominación de atrofias musculares espinales (AME) de la infancia, se agrupan una serie de procesos que tienen en común ciertas características, como ser determinadas genéticamente en forma autosómica recesiva y cursar con atrofia muscular por degeneración de las neuronas del asta anterior de la médula espinal y, en las formas más graves, de los núcleos de los últimos pares craneales.1,2 Constituyen la segunda causa de enfermedad autosómica recesiva letal, después de la fibrosis quística,3 con una incidencia mundial descrita entre 1/6000 y 1/10,000 nacimientos y una tasa de portadores entre 1/35 y 1/50.3,4 En México, la incidencia es de 0.5-1/25,000 nacimientos y solo existen reportes de casos aislados.5-9 El gen común para las formas infantiles se localiza en el cromosoma 5.10 Ha sido clonado e identificado su producto.11 Aunque el gen afectado es el mismo, desde el punto de vista clínico se consideran varias formas de la enfermedad, según la edad de inicio y evolución. Estas se mencionan a continuación:12,13
• Tipo I o enfermedad de Werdnig-Hoffmann. Es la forma más severa. Se inicia in útero o durante los primeros meses de vida. La muerte suele ocurrir antes de los dos años de edad.
• Tipo II o intermedia. Se presenta antes de los 18 meses de edad. La supervivencia de estos individuos depende del grado de complicaciones respiratorias. En esta forma clínica, con el abordaje actual (rehabilitación, cirugía de la escoliosis y, sobre todo, ventilación no invasiva) la supervivencia llega a alcanzar la edad adulta.
• Tipo III o enfermedad de Kugelberg-Welander. Se presenta después de los 18 meses de edad. En esta forma, la gravedad es muy variable porque depende de cuándo se inicia la enfermedad: antes o después de los 3 años de vida.
En 1978, argumentando la dificultad de precisar la edad de inicio y que la gravedad no siempre guarda relación con este dato -excepto para la enfermedad de Werdnig-Hoffmann-, V. Dubowitz propuso una clasificación eminentemente pragmática que, aunque también considera 3 formas, se basa en la capacidad del niño para mantenerse sentado, de pie o caminar sin ayuda. La primera es forma grave en la que el niño no logra el control cefálico y es incapaz de mantenerse sentado sin ayuda. En la forma intermedia, es capaz de mantenerse sentado sin ayuda pero no de mantenerse en pie ni caminar. En la forma leve, puede mantenerse en pie o caminar.14
Desde el punto de vista molecular, la causa es una mutación homocigota en el gen de sobrevida de la motoneurona (SMN) , que se ubica en el brazo largo del cromosoma 5 (5q11.1-13.3). Este gen está presente en múltiples copias en el genoma humano: una telomérica (SMN1) y varias copias centroméricas (SMN2) , que se diferencian en solamente cinco nucleótidos. El gen SMN2 presenta tendencia a un ensamblaje génico alternativo (alternative splicing) durante la transcripción del ARNm, que origina una proteína truncada que conserva solo 10% de la proteína SMN completa. Esta proteína normal no logra compensar la pérdida de la proteína por mutación del gen SMN1 .15,16 Las deleciones del exón 7 y 8 o solo la deleción 7 del gen SMN1 son responsables de más de 95% de los casos de AME.17 De este modo, la detección de una deleción homocigota de, por lo menos, el exón 7 de SMN1 , constituye una herramienta para el diagnóstico de AME que alcanza una sensibilidad cercana a 95% y una especificidad de 99%.17
Caso clínico
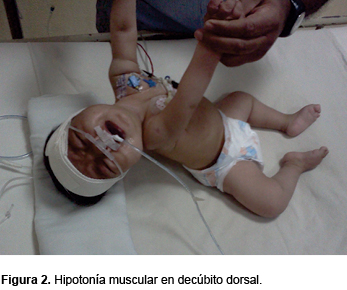
Se trata de una paciente de sexo femenino de 6 meses de edad, producto de G III, embarazo a término, obtenida por cesárea. Sin antecedentes de asfixia perinatal, con peso al nacimiento de 3200 g y talla de 49 cm. Fue alimentada exclusivamente al seno materno. Presenta cuadro de inmunizaciones completo para su edad. Padre de 32 años, madre de 30 años con antecedente de aborto espontáneo en G II. Ambos padres aparentemente sanos, procedentes de una comunidad pequeña y con los mismos apellidos, aunque sin parentesco. Un hermano de 5 años sano. Ingresó al Servicio de Urgencias con infección respiratoria aguda de 5 días de evolución, manifestada por rinorrea, tos y fiebre no cuantificada. A su ingreso presentó dificultad respiratoria grave, con broncoespasmo severo. Ameritó ventilación mecánica controlada. Se manejó con broncodilatadores, esteroides y relajantes musculares. Llamó la atención la dificultad para el retiro del ventilador ya que, después de haber suspendido el relajante muscular, la paciente no mostró un adecuado esfuerzo respiratorio. A la exploración física, la paciente presentó hipotonicidad de las cuatro extremidades con posición característica de las extremidades superiores, que consiste en brazos extendidos a lo largo del tronco, pronación de antebrazos, así como piernas abiertas y pegadas al plano de apoyo (Figura 1), con marcada hipotonía muscular (Figuras 2 y 3). La debilidad de la musculatura intercostal determinaba una respiración diafragmática que daba al abdomen la apariencia de un globo. La facies era hipomímica con mirada viva y alerta. Presentaba fasciculaciones de la lengua con discretos pliegues que le daban el aspecto cerebroide.


Como antecedente de importancia, el padre refirió la falta de sostén cefálico, así como cierta hipotonicidad. La madre refirió que, durante el embarazo, los movimientos fetales fueron adecuados.
Se solicitaron pruebas de PKC y aldolasa, que resultaron normales.
Se efectuó un estudio electromiográfico que reportó imágenes de inestabilidad de la membrana en 100% de los músculos estudiados, con abundantes fibrilaciones y ondas positivas. No se registraron fasciculaciones. Se obtuvieron escasos potenciales de unidades motoras, que reflejan la pérdida de las células del asta anterior de la médula espinal. El patrón de interferencia se encontró muy disminuido e incompleto, con un número muy limitado de potenciales y con mayor afectación en los músculos de miembros pélvicos. Se encontró ausencia de potenciales motores al efectuar la velocidad de conducción nerviosa en el nervio mediano y peroneo derecho. Se concluyó que el estudio fue anormal y compatible con polineuropatía motora con daño mielínico axonal severo, secundaria a atrofia muscular espinal infantil progresiva aguda.
Se realizó el análisis molecular del gen SMN-1 a partir del ADN genómico obtenido de leucocitos totales de sangre periférica. Se reportó la ausencia de los exones 7 y 8 correspondientes al gen SMN-1 , y la integridad de los exones 7 y 8 del pseudogen SMN-2 (Figura 4). Lo anterior indica un estado homocigoto para una deleción que compromete ambos alelos SMN-1 .
Discusión
La enfermedad de Werdning-Hoffmann es la forma más severa de las atrofias musculares espinales de la infancia, y se manifiesta en los primeros meses de vida, como en este caso. El diagnóstico de sospecha es fundamentalmente clínico, por lo que se debe pensar en atrofia muscular infantil ante un lactante con marcada hipotonía. Es importante considerar que la hipotonía puede pasar desapercibida para el familiar, por lo que la causa de la hospitalización podría ser una insuficiencia respiratoria, como en esta paciente, lo que se puede confundir con una neumonía si no se realiza un interrogatorio y la adecuada exploración física.
No debe olvidarse el abordaje de estudio de un paciente hipotónico, cuyos diagnósticos diferenciales pueden ser enfermedades metabólicas, por ejemplo enfermedad de almacenamiento de glucógeno, hipotiroidismo, intoxicaciones (botulismo) y enfermedades neuromusculares congénitas.
Para la confirmación diagnóstica es necesario realizar estudios neurofisiológicos, biopsia de músculo (en caso de duda) y, en la actualidad, el estudio molecular, que es el estándar de oro para el diagnóstico definitivo. En esta paciente no se realizó la biopsia muscular; sin embargo, el estudio de electromiografía fue compatible con atrofias musculares espinales (AME). El análisis molecular confirmó el diagnóstico clínico de atrofia muscular espinal, debido a un estado homocigoto para la deleción de los exones 7 y 8 del gen SMN-1 . El resultado del estudio molecular permite proporcionar asesoramiento genético a la familia, ya que indica una probabilidad mayor a 99% de que los padres biológicos de la paciente sean heterocigotos o portadores sanos de la deleción de los exones 7 y 8 correspondientes al gen SMN-1 . Así, al ser la AME una entidad con herencia autosómica recesiva, el riesgo de recurrencia de la enfermedad en la descendencia de los padres, independientemente del sexo, es de 25% en cada embarazo.
Los recientes progresos en la identificación de alteraciones moleculares en estos padecimientos, así como los avances en la tecnología médica, motivaron la creación de un comité multidisciplinario de expertos para el manejo de la AME. Después de analizar los estudios existentes, realizaron un consenso que se utiliza como guía para el cuidado de pacientes con atrofia muscular espinal, incluyendo el diagnóstico y nuevas intervenciones en el manejo de las complicaciones respiratorias, gastrointestinales, de nutrición, ortopédicas, de rehabilitación y, también, sobre los cuidados paliativos.18 Sin embargo, no existe tratamiento curativo para la enfermedad de Werdnig-Hoffman; solamente para la prevención y el manejo de las complicaciones. El apoyo ventilatorio es importante, así como el manejo de las infecciones.18,19 Se puede proporcionar manejo ventilatorio no invasivo,18,20 a pesar de que para esta forma clínica de AME no hay consenso a este respecto, y en la mayor parte de los centros no suele aplicarse este tipo de manejo ventilatorio.
Desafortunadamente, el pronóstico a corto plazo es malo. Nuestra paciente mejoró de su proceso respiratorio y, al ya no requerir del manejo ventilatorio, fue dada de alta por máximo beneficio. La madre otorgó el consentimiento informado para la publicación del caso clínico, incluyendo las fotografías de la paciente.
REFERENCIAS
1. Scheffer H. Spinal muscular atrophy. Methods Mol Med 2004;92:343-358. doi:10.1385/1-59259-432-8:343. [ Links ]
2. Iannaccone ST, Smith SA, Simard LR. Spinal muscular atrophy. Curr Neurol Neurosci Rep 2004;4:74-80. [ Links ]
3. Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 2005;48:885-896. [ Links ]
4. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371:2120-2133. doi:10.1016/S0140-6736(08)60921-6. [ Links ]
5. Garza-Alatorre AG, Rodríguez-Bonito R, López-Espinoza JA, Nieto-Sanjuanero A. Atrofia muscular espinal tipo I. Reporte de un caso atípico. Rev Mex Pediatr 2001;68:69-71. [ Links ]
6. Sánchez MJ, Moreno GAM, Romero BBL, Hernández AJ, García DC, Sánchez AA. Lactante con atrofia muscular espinal y encefalopatía hipóxico-isquémica. Bol Med Hosp Infant Mex 2010;67:63-73. [ Links ]
7. Palmer-Morales Y, Pacheco-Flores G, Ames-Guevara Y, Gaxiola-Apodaca M, Gaspar-Franco D, Landavazo-Acuña G, et al. Enfermedad de Werdnig-Hofmann. Dos casos clínicos. Rev Med Inst Mex Seguro Soc 2010;48:317-319. [ Links ]
8. Collado-Ortiz MA, Shkurovich-Bialik P, González de Leo S, Arch-Tirado E. Atrofia espinal tipo I (síndrome de Werdnig-Hoffmann). Reporte de un caso. Cir Cir 2007;75:119-122. [ Links ]
9. Padrón-Arredondo G, López-Gómez L. Atrofia muscular espinal infantil tipo I. Presentación de un caso presuntivo y revisión de la literatura. Salud Tab 2007;13:700-703. [ Links ]
10. Melki J, Abdelhak S, Sheth P, Bachelot MF, Burlet P, Marcadet A, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 1990;344:767-768. [ Links ]
11. Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155-165. [ Links ]
12. Brooke MH. A Clinician's View of Neuromuscular Diseases. London: Williams & Wilkins; 1985. pp. 36-80. [ Links ]
13. Munsat TL, Davies K. Workshop report. Spinal muscular atrophy. 32nd ENMC International Workshop. Naarden, The Netherlands, 10-12 March 1995. Neuromuscul Disord 1996;6:125-127. [ Links ]
14. Dubowitz V. Muscle Disorders of Childhood. Philadelphia: Saunders; 1978. pp. 146-190. [ Links ]
15. Burlet P, Bürglen L, Clermont O, Lefebvre S, Viollet L, Munnich A, et al. Large scale deletions of the 5q13 region are specific to Werdnig-Hoffmann disease. J Med Genet 1996;33:281-283. [ Links ]
16. Scheffer H, Cobben JM, Matthijs G, Wirth B. Best practice guidelines for molecular analysis in spinal muscular atrophy. Eur J Hum Genet 2001;9:484-491. [ Links ]
17. Ogino S, Wilson R. Spinal muscular atrophy: molecular genetics and diagnostics. Expert Rev Mol Diagn 2004;4: 15-29. [ Links ]
18. Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, et al. Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027-1049. [ Links ]
19. Castiglione C, Levicán J, Rodillo E, Garmendia MA, Díaz A, Pizarro L, et al. Atrofia muscular espinal: caracterización clínica, electrofisiológica y molecular de 26 pacientes. Rev Med Chile 2011;139:197-204. [ Links ]
20. Petrone A, Pavone M, Testa MB, Petreschi F, Bertini E, Cutrera R. Noninvasive ventilation in children with spinal muscular atrophy types 1 and 2. Am J Phys Med Rehabil 2007;86:216-221. [ Links ]

