










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.70 no.5 México sep./oct. 2013
CASO CLÍNICO
Síndrome de Ohtahara asociado con fístula traqueoesofágica en ''H''
Ohtahara syndrome associated with H-type tracheoesophageal fistula
Mario Eduardo Rodríguez Miralrío,1 Marco Antonio Toxtle Román,2 Carlos Javier Huesca Quintero1,3
1 Hospital General Regional 25 Zaragoza, Instituto Mexicano del Seguro Social, México, D.F.
2 Hospital Integral Atlapexco, SSA Atlapexco, Hidalgo.
3 Centro Médico Nacional La Raza, México D.F.
Autor de correspondencia:
Dr. Marco Antonio Toxtle Román
Correo electrónico: drmatr@hotmail.com
Fecha de recepción: 03-03-13
Fecha de aceptación: 12-06-13
Resumen
Introducción. El síndrome de Ohtahara es una encefalopatía epiléptica infantil temprana, caracterizada por espasmos tónicos frecuentes, crisis parciales y, ocasionalmente, mioclonías. El electroencefalograma interictal muestra un patrón característico de descargas de puntas que alternan con fases de supresión de la actividad eléctrica cerebral (brote-supresión). Las manifestaciones clínicas inician habitualmente antes de los 20 días de vida extrauterina. Los pocos casos reportados en la literatura no mencionan malformaciones congénitas asociadas.
Caso clínico. Documentamos el primer caso clínico de un lactante masculino de 6 meses de edad con síndrome de Ohtahara asociado con una fístula traqueoesofágica tipo ''H''.
Conclusiones. La asociación del síndrome de Ohtahara con la fístula traqueoesofágica pareciera deberse a una presentación fortuita y no con relación al síndrome neurológico.
Palabras clave: síndrome de Ohtahara, convulsiones, brote-supresión, fístula traqueoesofágica.
Abstract
Background. Ohtahara syndrome is an early infantile epileptic encephalopathy characterized by frequent tonic spasms, partial seizures and occasional myoclonus. Interictal EEG characteristically shows a pattern of burst of spikes alternating with phases of suppression of brain electrical activity (''burst-suppression''). Clinical manifestations usually begin before 20 days of life. The few cases reported in the literature do not mention associated congenital malformations.
Case report. We report the first case of a 6-month-old male infant with Ohtahara syndrome associated with H-type tracheoesophageal fistula.
Conclusions. The association between Ohtahara syndrome and tracheoesophageal fistula may be due to a fortuitous presentation without any relationship with the neurological syndrome.
Key words: Ohtahara syndrome, seizures, burst-suppression, tracheoesophageal fistula.
Introducción
El síndrome de Ohtahara (SO) o encefalopatía epiléptica infantil temprana (EIEE) es un trastorno neurológico progresivo y debilitante que resulta en convulsiones intratables y retraso mental severo. Clínicamente, el SO se caracteriza por espasmos tónicos iniciales asociados con un patrón severo y continuo de la actividad de ráfaga.1 Su etiología es idiopática o sintomática.2 Fue descrito por primera vez, en 1976, por Ohtahara, como un síndrome caracterizado por espasmos tónicos que ocurrían antes de los 20 días de vida, generalmente en los primeros cinco días, y que carecían de los mioclonos fragmentarios o crisis clónicas descritas por Aicardi y Gutiérrez, pero que mostraba el mismo tipo de patrón electroencefalográfico (EEG) de brote-supresión.3-5
Por otra parte, la fístula traqueoesofágica (FTE) es una malformación congénita que ocurre como consecuencia de una falla en el curso del desarrollo del tabique esofágico respiratorio (canal traqueobronquial) dentro del intervalo de la cuarta a la sexta semana de desarrollo embrionario. Presenta una incidencia actual de 1:4000 recién nacidos, afectando por igual a ambos sexos. En 50-70% de los casos se asocia con anomalías cardiacas, gastrointestinales y genitourinarias.6
No se han reportado casos en la literatura de pacientes con estas dos entidades, dado que tienen un origen embriológico distinto cuya formación comprende diferentes edades gestacionales.
En este trabajo se documentó la presencia de estas dos anormalidades en el mismo paciente.
Caso clínico
Lactante masculino de 6 meses, producto de G III de madre de 32 años, quien cursó con embarazo normoevolutivo y control prenatal adecuado. Fue obtenido en medio hospitalario a las 37 semanas de gestación por vía vaginal, con un peso al nacer de 2,055 g y talla de 48 cm. Apgar desconocido. Fue egresado a su domicilio en binomio a las 24 horas después de nacer. Sin antecedentes heredo-familiares de crisis convulsivas o epilepsia. A las 72 horas de vida extrauterina presentó paro respiratorio secundario a crisis convulsivas, por lo que se trasladó a su unidad hospitalaria más cercana. A la exploración física se encontró hipotrófico, hipoactivo, con espasticidad global. Cráneo microcéfalo con cabalgamiento de suturas y fontanela anterior amplia. A nivel cardiopulmonar sin alteraciones aparentes, con pobre tolerancia a la vía oral, sin reflejo de succión. Las extremidades mostraban hiperreflexia y clonus agotable. Se decidió manejo avanzado de la vía aérea por crisis convulsivas recurrentes y refractarias a tratamiento con midazolam 100 μg/kg/dosis y difenilhidantoína a 7 mg/kg/día, por lo que se realizó referencia a tercer nivel para protocolo de estudio.
El paciente persistió con crisis convulsivas tónico-clónicas (Figura 1), por lo que se inició protocolo de estudio. Se realizó tamiz metabólico ampliado en el que no se identificaron alteraciones o errores innatos del metabolismo. Cultivos citológico, citoquímico y de líquido cefalorraquídeo, normales. Tomografía de cráneo con leucoencefalomalacia global, probablemente secundaria a encefalopatía hipóxico-isquémica (Figura 2). Al realizar EEG, se identificó patrón de brote-supresión (BS), por lo que se estableció el diagnostico de SO por servicio de neurología pediátrica (Figura 3). Se inició tratamiento con vigabatrina a 80 mg/kg/día y con ácido valproico a 20 mg/kg/día, logrando el control de las crisis convulsivas. Se refirió a un hospital de segundo nivel para continuar su manejo.

Al reingreso presentó succión débil, por lo que se realizó gastrostomía para mejorar la nutrición y, a través de una serie esofagogastroduodenal, se identificó el paso del medio de contraste a vía aérea (Figura 4), diagnosticándose la presencia de fístula traqueoesofágica en ''H'' (Figura 5).
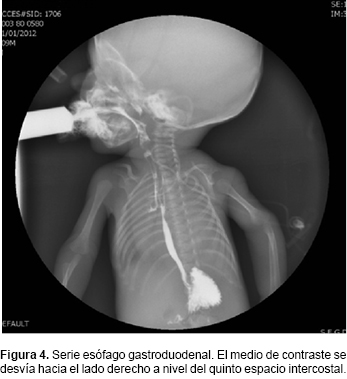
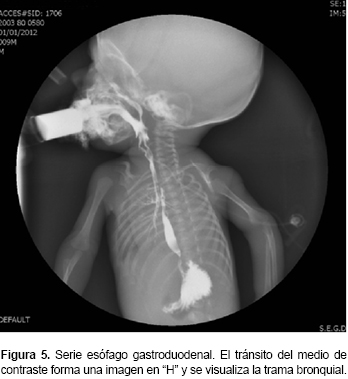
Durante su estancia hospitalaria el paciente cursó con infección nosocomial por Pseudomonas aeruginosa (hemocultivo central y periférico positivos). El antibiograma reportó sensibilidad intermedia a meropenem (120 mg/kg/día) y cefepime (150 mg/kg/día). Sin embargo, presentó pobre respuesta al tratamiento antibiótico, y desarrolló choque séptico con coagulación intravascular diseminada, lo que finalmente ocasionó su defunción a los 6 meses de vida.
Discusión
El SO es una entidad epiléptica que, como el síndrome de West (SW), puede ser idiopático o sintomático y es dependiente de la edad.7,8
Las características más frecuentes de este síndrome son su comienzo antes de los 3 meses de vida, un mal pronóstico, refractariedad al tratamiento antiepiléptico y el patrón EEG de BS.8-10 Este patrón EEG es la característica más constante y de mayor especificidad. Ohtahara describió los parámetros utilizados para diagnosticar el síndrome:
1) Brotes de una amplitud de 150 a 300 μV de 3 seg de duración y frecuencia mixta, que incluyen descargas en punta
2) Períodos de supresión y una amplitud reducida con duración de 5 a 7 seg11
Los niños con SO presentan cuatro características comunes, aunque carentes de especificidad:
1) Comienzo de las convulsiones en la primera infancia
2) Mal pronóstico
3) Convulsiones tónicas espasmódicas
4) Actividad de supresión y salva en el EEG
El período de recién nacido es una etapa de la vida en la cual las convulsiones son frecuentes, y regularmente se asocian con un mal pronóstico.11
La diferencia entre el síndrome de Ohtahara y la encefalopatía mioclónica temprana (EMT) ha sido enfatizada por algunos autores, quienes clasifican a todo recién nacido con convulsiones mioclónicas multifocales como EMT. Ambos síndromes comparten el patrón de BS en el EEG, independientemente del estado de sueño-vigilia.8 Estos autores se basan en la presencia de mioclonías para excluir el síndrome de Ohtahara.9,11
Sin embargo, el BS en EMT es generalmente más evidente durante el sueño, por lo que los EEG de grabación durante el sueño son de la mayor importancia en el diagnóstico. Cabe destacar que el BS en EIEE se encuentra desde el inicio solamente en casos limitados (que, por lo general, desaparecen al cabo de seis meses), mientras que en EMT se encuentra persistentemente en todos los casos, incluso después de 1 año de edad y hasta el final del seguimiento.1
Actualmente, se incluyen como dos síndromes separados en la clasificación de epilepsias, pero se utilizan indistintamente como sinónimos en el catálogo McKusick (OMIM).1,12
Estudios moleculares preliminares parecen indicar diferencias genéticas entre las dos entidades (EMT es probablemente causado por la ruptura de la neurogulina-1 receptor ErbB4).13 A pesar de una serie de coincidencias clínicas y neurofisiológicas,14 EIEE, EMT y otras encefalopatías epilépticas primarias como SW y síndrome de Lennox-Gastaut son probablemente parte de un fenotipo continuo, cuyas diferencias clínicas (por ejemplo tipo de crisis, los patrones de EEG, la transición a otras formas de epilepsia, historia natural y la respuesta al tratamiento) son dictadas por proteínas anormales como resultado de mutaciones genéticas específicas.1
Basados en el cuadro clínico y en el patrón EEG de BS, este paciente presentó SO. Las malformaciones asociadas más frecuentemente con este síndrome son las del SNC. Además, este caso estaba asociado con FTE, el cual se encontró de manera fortuita.
La mayor parte de las características originales propuestas por Ohtahara han sido consistentes a través del tiempo.15-17 Otras características neurofisiológicas y hallazgos etiológicos han sido integradas como resultado de los avances más recientes.18 El SO es raro, desde un punto de vista epidemiológico. Los datos son escasos y controvertidos, con una prevalencia que va desde 0.2 a 4% de las epilepsias en la infancia.1,19
Cuadro clínico del síndrome de Ohtahara
La edad de aparición de las convulsiones se limita a la etapa neonatal o periodos infantiles muy tempranos. En un estudio, en aproximadamente 30% de los casos se manifestaron las convulsiones dentro de los primeros 10 días de vida, en comparación con aproximadamente 70% de los niños de un mes de edad.18 El patrón principal de las convulsiones es de espasmos tónicos, con o sin agrupamiento, que se observa en todos los casos; puede ser generalizado y simétrico o lateralizado. La duración de los espasmos es corta (hasta 10 seg). Estos espasmos son clínicamente similares a los del SW, aunque con algunas características diferentes: aparecen durante el estado de vigilia y sueño y sin agrupamiento.18,20-22
Además de los espasmos tónicos, se observan en aproximadamente el 30% de los casos convulsiones parciales motoras, hemiconvulsiones o convulsiones generalizadas tónicas. La frecuencia diaria de ataques es muy alta. Va de 100 a 300 episodios en niños con convulsiones aisladas, y de 10 a 20 agrupaciones en individuos que experimentan los grupos de convulsiones. Las crisis mioclónicas raramente se registran.1,17
Hallazgos del EEG interictales
La característica más específica del EEG es el patrón BS, que presenta brotes de alto voltaje de ondas lentas mezcladas con puntas multifocales, alternando con un trazo plano (isoeléctrico) de supresión a una velocidad aproximadamente regular. Entre otras características distintivas se encuentra la apariencia consistente, tanto en estado de vigilia como en sueño, y la periodicidad. Esta es un hallazgo muy específico y de diagnóstico.20,21
Electroencefalográficamente, el patrón de BS disminuye gradualmente a partir de los 3 meses de edad. Por lo general desaparece a los 6 meses: se transforma en hipsarritmia en la mayoría de los casos (entre los 2 y 6 meses de edad) o muestra una transición más lenta y se transforma en punta-ondas o múltiples picos independientes.23
Hallazgos de neuroimagen
Las anormalidades estructurales más comunes incluyen asimetría cerebral, macrocefalia, hemimegalencefalia, paquigiria agiria, polimicrogiria, displasia cortical focal, displasia olivar, disgenesia del collicoli y anomalías de la fosa posterior.22,24-26 Sin embargo, la anomalía cerebral más común es la hipodesmielinización y la atrofia cortical difusa.
Estrategias terapéuticas
La administración de ACTH (hormona adenocorticotropa) ha demostrado ser eficaz en el SO y, en algunos casos, incluso después de la transición a SW. También se han reportado casos anecdóticos de eficacia con clonazepam y acetazolamida. Se han utilizado dosis altas de vitamina B6, valproato de sodio, vigabatrina o una dieta cetogénica, pero los resultados no han sido buenos. En un estudio reciente se ha utilizado la zonisamida para suprimir las convulsiones. Sin embargo, estos resultados deben interpretarse con precaución. La hemisferectomía en niños seleccionados con anormalidades cerebrales (por ejemplo, macrocefalia o calloso) ha demostrado resultados prometedores, lo que sugiere que la edad temprana no es contraindicación para la cirugía.1
Al parecer, la asociación entre SO y FTE se debe a una situación fortuita, y no existe relación con el síndrome neurológico. Este caso se manejó con vigabatrina y ácido valproico, con lo que se logró el control de las convulsiones, aunque lo ideal es un monitoreo EEG continuo para una adecuada evidencia clínica de respuesta al manejo antiepiléptico.
Agradecimientos
Dr. Daniel San Juan Orta (Neurólogo), C.P. Abigail Silva Hernández y Dra. Ariadna Martínez Rivas
REFERENCIAS
1. Pavone P, Spalice A, Polizzi A, Parisi P, Ruggieri M. Ohtahara syndrome with emphasis on recent genetic discovery. Brain Dev 2012;34:459-468. [ Links ]
2. Campistol J. Convulsiones neonatales refractarias. Medicina (Buenos Aires) 2009;69:41-50. [ Links ]
3. Faúndez JC. Convulsiones neonatales. Rev Pediatr Elec 2005;2:26-35. [ Links ]
4. Feld V, Vita C. Encefalopatía epiléptica infantil temprana (EEIT). Manifestación clínica, etiología y tratamiento neonatal, a propósito de un caso. Rev Hosp Mat Inf Ramón Sardá 2011;30:54-57. [ Links ]
5. Palencia R. Síndromes convulsivos en el periodo neonatal. Bol Pediatr 2002;42:31-39. [ Links ]
6. Instituto Nacional de Perinatología. Defectos del aparato digestivo. Atresia de esófago y fistula traqueoesofágica. En: Normas y Procedimientos de Neonatología. México D.F.: InPer; 2009. pp. 120-123. [ Links ]
7. Aviña-Fierro JA, Hernández-Aviña DA. Encefalopatía epiléptica infantil temprana. Descripción de un caso de síndrome de Ohtahara. Rev Mex Pediatr 2007;74:109-112. [ Links ]
8. Krasemann T, Hoovey S, Uekoetter J, Bosse H, Kurlemann G, Debus OM. Early infantile epileptic encephalopathy (Ohtahara syndrome) after maternal electric injury during pregnancy: etiological considerations. Brain Dev 2001;23: 359-362. [ Links ]
9. Aicardi J. Early myoclonic encephalopathy. En: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. London: John Libbey Eurotext; 1985; pp. 12-21. [ Links ]
10. Korff CM, Vulliemoz S, Picard F, Fluss J. Ohtahara syndrome or early-onset West syndrome? A case with overlapping features and favorable response to vigabatrin. Eur J Paediatr Neurol 2012;16:753-757. [ Links ]
11. Yelin K, Alfonso I, Papazian O. Sindrome de Ohtahara. Rev Neurol 1999;29:340-342. [ Links ]
12. OMIM. Online Mendelian Inheritance in Man. Baltimore: Johns Hopkins University Press; 2008. [ Links ]
13. Guerrini R. Epilepsy in children. Lancet 2006;367:499-524. [ Links ]
14. Backx L, Ceulemans B, Vermeesch JR, Devriendt K, Van Esch H. Early myoclonic encephalopathy caused by a disruption of the neuregulin-1 receptor ErbB4. Eur J Hum Genet 2009;17:378-382. [ Links ]
15. Ohtahara S, Ishida T, Oka E, Yamatogi Y, Inoue H, Kanda S. On the specific age-dependent epileptic syndrome: the early-infantile epileptic encephalopathy with suppression-burst. No To Hattatsu 1976;8:270-280. [ Links ]
16. Ohtahara S. A study of the age-dependent epileptic encephalopathy. No To Hattatsu 1977;9:2-21. [ Links ]
17. Delgado-Ochoa MA, Marca-González SR, Huerta-Hurtado AM, Pérez-Ramírez JM, Hernández-Hernández M, Barragán-Pérez EJ. Síndrome de Ohtahara: casuística de 10 años en el Hospital Infantil de México Federico Gómez. Rev Med Hondur 2007;75:182-185. [ Links ]
18. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782-788. [ Links ]
19. Thambyayah M. Early epileptic encephalopathies including West syndrome: a 3-year retrospective study from Klang Hospital, Malaysia. Brain Dev 2001;23:603-604. [ Links ]
20. Ohtahara S, Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol 2003;20:398-407. [ Links ]
21. Fusco L, Pachatz C, Di Capua M, Vigevano F. Video/EEG aspects of early-infantile epileptic encephalopathy with suppression-bursts (Ohtahara syndrome). Brain Dev 2001;23:708-714. [ Links ]
22. Beal JC, Cherian K, Moshe SL. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 2012;47:317-323. [ Links ]
23. Ohtahara S, Ohtsuka Y, Yamatogi Y, Oka E. The early-infantile epileptic encephalopathy with suppression-burst: developmental aspects. Brain Dev 1987;9:371-376. [ Links ]
24. Bastos H, Sobral da Silva PF, Veloso de Albuquerque MA, Mattos A, Santos RR, Ohlweiler L, et al. Proteus syndrome associated with hemimegalencephaly and Ohtahara syndrome: report of two cases. Seizure 2008;17:378-382. [ Links ]
25. Hmaimess G, Raftopoulos C, Kadhim H, Nassogne MC, Ghariani S, de Tourtchaninoff M, et al. Impact of early hemispherotomy in a case of Ohtahara syndrome with left parieto-occipital megalencephaly. Seizure 2005;14: 439-442. [ Links ]
26. Raspall M, Ortega-Aznar A, del Toro M, Roig M, Macaya A. Neonatal rigid-akinetic syndrome and dentato-olivary dysplasia. Pediatr Neurol 2006;34:132-134. [ Links ]
Nota
Este artículo puede ser consultado en versión completa en: http://www.medigraphic.com/BMHIM

