











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
Rhabdomyosarcoma (RMS) is a malignant tumor of striated muscle of mesenchymal origin regarded as the most recurrent soft tissue sarcoma in children and adolescents, with an annual incidence of 4.3 cases per million. Approximately two-thirds of the cases are diagnosed in children (typically around 6 years of age), and are slightly more common in males, with a male/female ratio of 1.4:1.1 Due to its origin in a totipotent cell, rhabdomyosarcomas not only occur in skeletal muscle but other locations, such as the head, neck, genitourinary tract and bile ducts. Approximately 40% of RMS occur in the region of the head and neck, 20% in the genitourinary region, 20% in the extremities and the remaining 20% in other sites.1
The embryonal and alveolar variants are the more frequent histological types, comprising 70 to 20% of the cases, respectively.2 The embryonal rhabdomyosarcoma (ERMS) is the most common subtype in infants and young children and represents more than two-thirds of all the RMS.1,3 This tumor is composed of a soft mixture of fusiform cells with areas of soft, loose stroma, and it is associated with loss of heterozygosity at the 11p15 locus.3 The tumors that are located in the head, neck, and genitourinary region often present this histological type.
Alveolar rhabdomyosarcomas (ARMS) usually appear in adolescence; they are typically located in the extremities and have a high capacity to metastasize.1,4 Their histology is characterized by a septum of fibrous connective tissue with neoplastic cells attached (Figure 1A), similar to the alveolar spaces observed in the lung, where some of the cells detach and occupy the space. It is composed of cells uniformly polygonal with high grade round or oval hyperchromatic nucleus (Figure 1B).1 Unlike the ERMS, the ARMS exhibit two particular types of chromosomal translocations: between chromosomes 2 and 13, t (2;13) (q35;q14), and between chromosomes 1 and 13, t (1;13) (p36;q14), which occur in 80% of the cases.4 These genetic alterations lead to the fusion of two families of transcription factors. The first is located on chromosome 1 or 2 and involves transcription factors PAX3 and PAX7, respectively. PAX family of transcription factors is a group of genes involved in the differentiation of organs and tissues, and possess an N-terminal DNA binding domain, which includes a paired box, homeobox motifs, and a C-terminal trans-activation domain, while the second class involves members of the family of the forkhead (FKHR) transcription factors or FOXO1.

Figure 1 (A) Histological appearance of alveolar rhabdomyosarcoma: fibrous connective septa are forming pseudo-alveolar structures, in which neoplastic cells are embedded. (B) In some areas, they occupy the entire space forming a solid neoplasm.
The transcription factors of PAX and FOXO1 families possess an N-Terminal domain of union with the DNA and a C-terminal domain of transactivation. The breaking points for PAX and FOXO1 occur at introns 7 and 1, respectively. Fused genes codify for two chimeric proteins with oncogenic activity, PAX3/FOXO1, and PAX7/FOXO1. These proteins are composed of a 5′ domain of union to the DNA (PAX) and a 3′ transactivation domain (FOXO1).4 It has been demonstrated that PAX3/FOXO1 and PAX7/FOXO1 transcripts are present in 55 and 22% of the ARMS, respectively, while the remaining ARMS are negative for the fusion gene.5 It is known that ARMS with the PAX7/FOXO1 translocation have a much more favorable prognosis compared to those who carry the translocation PAX3/FOXO1. The median 4-year survival for the former is 75% and 8% for the latter.6 The patients with ARMS who often have metastasis at diagnosis have a short median survival. In addition, the presence of proteins of the fusion gene PAX3/7-FOXO1 is associated with an unfavorable prognosis.2
2. Clinical case
A male patient of two years and three months of age, native of Yucatan, who had no significant past medical history until five months of age when a violet bulk appeared in his left nostril. According to the parents, he was treated with chemotherapy and radiotherapy in a local clinic, with remission of the tumor two months later. When the boy was two years old, the tumor appeared again, so they came to our hospital.
On physical examination, his weight was 10.4 kg and his height 100 cm. He was awake, his right eye had normal pupil light reflex, but the left eye was not assessable because of a tumor located in the middle of the face, predominantly on the left side, purplish, fetid, fixed to deep planes. Teeth were displaced. He had no lymphadenopathies in the neck. No abnormalities were found in the thorax, abdomen or limbs.
Computed tomography (CT) showed a lobed tumor, with well-defined borders which involved from the frontal region up to the maxillary floor. Histopathological diagnosis was stage IV alveolar rhabdomyosarcoma with infiltration to bone marrow and cerebrospinal fluid. He was managed with antibiotics and began chemotherapy with adriamycin, actinomycin, cyclophosphamide, and vincristine. The first cycle was completed, and he was discharged. He received four cycles of chemotherapy, with a tumor size reduction of 50%. In the last cycle, cisplatin and irinotecan were added to the chemotherapeutic regime. The last CT scan showed widening of the nasal cavity and maxillary antrum with deformity of the left side of the face, displacement of the eyeball, infiltration to the medial wall of the orbit, poorly defined turbinates, medial and lateral walls of the left maxillary antrum and occupation of sphenoid sinuses.
The fourth chemotherapeutic cycle was suspended because of fever and neutropenia, abdominal bloating and decreased peristalsis because of hypokalemia and metabolic ileus. A gram-negative bacillus was isolated in blood culture. A nasogastric tube was placed, and neutropenic colitis was discarded. Respiratory insufficiency led to invasive mechanical ventilation. The patient had electrolyte imbalance and refractory septic shock, and finally died.
3. Discussion
Rhabdomyosarcoma is the most common soft tissue sarcoma in children; it is classified in embryonal rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma (ARMS), botryoid rhabdomyosarcoma and spindle cell rhabdomyosarcoma, with different phenotypes and clinical characteristics (Figure 2). Less common variants have been recently described, like sclerosing rhabdomyosarcoma and those with rhabdoid features. The ERMS and ARMS are the most prevalent and comprehend 70% and 20% of cases, respectively. Of these, ARMS is the one with the worse prognosis.2,3 This behavior has been associated with the expression of oncogenic fusion proteins resulting from chromosomal translocations, a tumorigenesis mechanism common with many types of cancer, including a third of the sarcomas.5

Figure 2 (A) Immunohistochemical staining showing positivity for desmin. An intermediate filament is present in the cytoplasm of striated muscle cells associated with Z bands. (B) Intense nuclear positivity for myoglobin, an hemeprotein present in skeletal muscle and serves as an oxygen carrier and reservoir; it appears late on the sequential muscle maturation and is positive in 95% of cases of rhabdomyosarcomas.
The most common chromosomal translocation found in ARMS is t(2;13) (q35;q14) PAX3/FOXO1, and the less frequently found is t(1;13) (p36;q14) PAX7/FOXO1.6,7 The translocations t(2;13) and t(1;13) result from the breakdown of specific genes that are within the chromosomic region 2q35 and 1p36, respectively, followed by fusion. The involved genes in chromosome 2 and 1 are PAX3 and PAX7, respectively, which encode for transcription factors of the paired box family.3 The genes that fuse with PAX3 and PAX7 are FOXO1 or FKHR, which are located on chromosome 13 and are members of the forkhead family of transcription factors. The fusion of these genes leads to the expression of fusion proteins, which act as transcriptional activators that contribute to tumor development by altering pathways of cell growth and apoptosis, modulating the myogenic differentiation, and stimulating motility and other metastatic pathways.3,8 During normal development, PAX3 expression is required for the migration of the precursors of skeletal muscle cells towards the extremities, while the expression of PAX7, a marker of satellite cells of skeletal muscle in adults, is required for the normal cell turnover. Both proteins are rapidly degraded during early myogenic differentiation. However, in the ARMS, the fusion of PAX3/PAX7 with FOXO1 gives them a longer half-life.7
RT-PCR and fluorescence in situ hybridization are methods that have been used to analyze the frequency of the fusion of PAX3 and PAX7 in ARMS (Figure 3). The Intergroup Rhabdomyosarcoma Study (IRS), which included 78 cases of ARMS, found that 77% of the cases were positive for the fusion, 55% of the ARMS expressed PAX3/FOXO1, 22% expressed PAX7/FOXO1, and 23% were negative for the fusion.9 In a study published in 2010, we found a similar trend: in 25 ARMS cases we found that 50% were positive for PAX3/FOXO1, 3% were positive for PAX7/FOXO1, and 30% were negative for the fusion gene.10 In this study, 37 ARMS cases were analyzed, 63% of which were positive for PAX3/FOXO1, 5% were positive for PAX7/FOXO1, and 32% were negative for the fusion, which is consistent with other reports. The detection of the oncogenic fusion proteins, particularly PAX3/FOXO1, has a significant prognostic value since the biological tumor behavior in RMS with this translocation is more aggressive, with poor response to treatment and frequent recurrences. In contrast, tumors with the PAX7/FOXO1 translocation have a better response to treatment, a usually lower clinical stage, and a longer survival.11
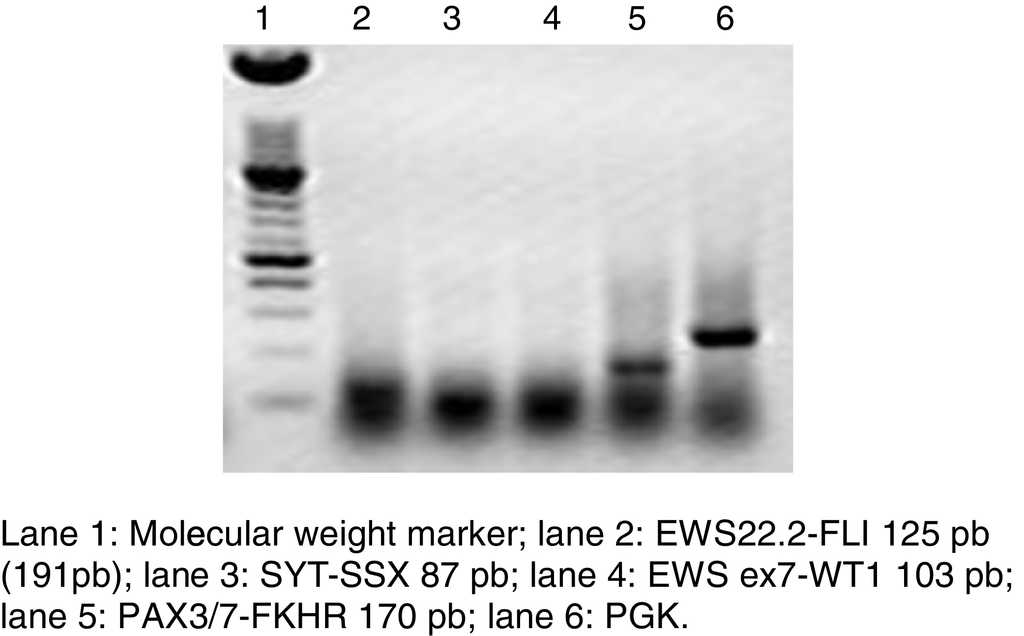
Figure 3 Agarose gel electrophoresis of an RT-PCR, where the representative translocation PAX3/7-FKHR of alveolar rhabdomyosarcoma is shown, discarding other solid neoplasms.
The mechanisms by which the chimeric protein PAX/FOXO1 contributes to oncogenesis of the RMS have been deeply studied. Apparently, the quantity and cellular localization of the protein are critical for its oncogenic activity. Both PAX3/FOXO1 and PAX7/FOXO1 have a transcriptional activity 100 times greater than the wild proteins PAX3 and PAX7.8,9 The proteins PAX/FOXO1 are expressed by themselves at high levels. The overexpression of PAX3 results from an increase in the transcription that is independent of the number of copies, while the high expression of PAX7 is associated with gene amplification.12,13 Aside from its overexpression, PAX3/FOXO1 is significantly more stable than PAX3, which quickly suffers proteolysis during muscle differentiation.14 Within the cell, these chimeric proteins can be found in the nucleus or the cytoplasm. In normal conditions, the location of the wild FOXO1 is controlled by AKT. When AKT is stimulated, it phosphorylates FOXO1, causing its retention in the cytoplasm. However, in ARMS, the fusion protein PAX/FOXO1 is resistant to AKT activity and remains predominantly in the nucleus.15 Another way in which the protein PAX/FOXO1 contributes in the oncogenesis is by preventing the tumor cell apoptosis through the expression of anti-apoptotic genes as Bcl-XL.
The main problem in the biology of rhabdomyosarcomas is their susceptibility to differentiate into skeletal muscle indefinitely, which is the result of alterations in the myogenic program, at the kinases (i.e., p38 MAPK) and the epigenetic level. Within the latter, there are the polycomb-repressive complex or JARID2, and the microRNAs (miRNAs).16 miRNAs are a family of non-codifying RNAs that regulate gene expression at the post-transcriptional level via the inhibition or degradation of messenger RNA. Currently, there is a significant number of miRNAs known to be involved in the induction and progression of rhabdomyosarcomas. The expression of several miRNAs is induced during the myogenic process, and their potential targets are genes that control the proliferation and differentiation of myoblasts, resulting in a deregulation of cell proliferation or an aberrant myogenic differentiation. The most studied miRNAs in skeletal muscle are miRNA-1, miRNA-133a/b, and miRNA-206, which are muscle specific, and miRNA-29b/c which is expressed ubiquitously.17
There is evidence pointing toward the family of miRNA-29 as tumor suppressors since aberrant expression of members of this family has been observed in several types of cancer. The miRNA-29s family is a preserved family, including miRNA-29a/b/c, which has an important role in cell proliferation, apoptosis, migration and invasion.17,18,19 miRNA-29b is the member of this family that is expressed at higher levels in normal conditions.19
The prognosis of patients with RMS depends on the grade of the tumor, age, type of resection, histology, presence of the mentioned translocations and number of sites with metastases.
The epithelial-mesenchyme transition (EMT) is an important event for the invasion and metastasis of the tumor, and it is associated with a poor prognosis and with chemoresistance. The EMT is a process in which the epithelial cells lose their polarity and the cell-cell adhesion, and can migrate and invade other organs.20 This process starts with the dissociation of the intercellular junctions (claudin, occludin, ZO-1, E-cadherin, and desmoplakin), resulting in the loss of the microvilli and apical-basolateral polarity.
The cells acquire an elongated morphology, increase the expression of smooth muscle α-actin, and enhance their ability to migrate. In the last stage of the EMT, cells acquire the potential to degrade the basement membrane through the expression of matrix metalloproteinases (MMP).20 The EMT is accompanied by molecular changes such as low expression of cytokeratin and vimentin, N-cadherin overexpression and the Snail transcription factor (E-cadherin expression inhibitor).20
Chemoresistance is one of the main problems in almost all types of cancer. Despite the fact that there is an improvement in the chemotherapeutic agents, many patients with cancer die because they develop chemoresistance. Several studies indicate that the miRNAs are involved in chemoresistance,21 including miRNA-29b.
The type of chemotherapy that patients receive with RMS depends on the risk factors they present. Low or intermediate risk patients receive vincristine, dactinomycin, and cyclophosphamide.22,23 In some intermediate risk patients, the intensification of the cyclophosphamide dose after total resection gives good results; however, in other patients, the intensification of chemotherapy does not improve the result.24 In high-risk patients, the combination of ifosfamide-etoposide or ifosfamide and doxorubicin is used. These patients usually have a poor prognosis. Interestingly, there has been a good response to the treatment in patients with high-risk ERMS with one or more metastases. This type of RMS has a better prognosis than other metastatic RMS.25 The fusion-negative ARMS behaves in a similar way to the ERMS.7 We have found that the expression of miRNA-29b is greater in the fusion-negative tumors, and they have a better response to chemotherapy.
Ethical disclosure
Protection of human and animal subjects. The authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of data.The authors declare that no patient data appear in this article.
Right to privacy and informed consent. The authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.

