











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Existe consenso en que la resucitación hemodinámica en el niño con choque séptico debe ser precoz y protocolizada1. Asimismo, la correcta y oportuna reanimación macrocirculatoria y microcirculatoria es de vital importancia para el pronóstico2-4.
El síndrome de disfunción multiorgánica (SDMO) se caracteriza por la falla simultánea de dos o más órganos o sistemas5. El desarrollo de SDMO es común en los pacientes ingresados en la unidad de cuidados intensivos (UCI). Dependiendo de la población estudiada, los criterios diagnósticos empleados y el fenotipo analizado en los pacientes con sepsis, la incidencia y la mortalidad son variables. El estudio SPROUT6 reportó un 25% de mortalidad hospitalaria por sepsis grave; de esta cohorte, el 58% presentó SDMO en el día de su reconocimiento, y el 40% falleció durante la hospitalización o desarrolló un nuevo o progresivo SDMO dentro de la semana siguiente. Villeneuve, et al.7 monitorizaron prospectivamente la ocurrencia diaria de SDMO en 842 pacientes de UCI. Según los criterios diagnósticos de Proulx y los de Goldstein, estos autores reportaron la existencia de SDMO en el 21.4% y el 37.3%, respectivamente. Sin embargo, la proporción de pacientes con SDMO al momento de su admisión y que fallecieron dentro de los 90 días siguientes fue mayor en los diagnosticados con los criterios de Proulx. En fecha más reciente, un estudio nepalés encontró SDMO en el 51% de los pacientes admitidos en la UCI8.
En ocasiones, a pesar de obtener adecuadas metas de resucitación (hemodinámicas y metabólicas) y de un oportuno soporte vital, muchos pacientes con sepsis desarrollan SDMO y fallecen9-13. Esto sugiere la participación de otros mecanismos fisiopatológicos14,15, como la hipoxia citopática o, más precisamente, la disoxia citopática, que se origina por un desacoplamiento del sistema de producción energética celular (fosforilación oxidativa)16,17. Se ha propuesto, a modo de hipótesis, que la disfunción mitocondrial es una alteración relevante en el desarrollo de la falla orgánica inducida por sepsis, aunque no se encuentra totalmente caracterizada y su presencia no es necesariamente evidencia de causalidad14. No obstante, los trastornos metabólicos descritos sugieren que la disfunción mitocondrial podría ser un mecanismo involucrado18. Asimismo, la determinación de su papel (patogénico o adaptativo) —es decir, la supresión de actividades dependientes de energía en favor de otras esenciales para la sobrevida celular— es controversial19,20. Actualmente existe evidencia clínica de una mayor deficiencia bioenergética21,22 en los pacientes con sepsis que fallecen, lo que sugiere que la disfunción mitocondrial es un mecanismo fisiopatológico trascendente que, además, explicaría la presencia de fallas orgánicas en el paciente con choque séptico.
La terapia orientada a la disfunción mitocondrial («resucitación metabólica») parece una opción razonable, posible y promisoria para la prevención y el tratamiento del SDMO23-25.
El objetivo de la presente revisión es actualizar los conocimientos respecto a las alteraciones mitocondriales, el papel de su adaptación y, finalmente, realizar algunas breves consideraciones referentes a la resucitación mitocondrial en el paciente séptico con SDMO.
Mitocondrias y respiración celular
Las mitocondrias, presentes en todas las células eucariotas, son vestigios de un proceso ancestral endosimbiótico eubacteriano (α-proteobacterias) ocurrido hace más de un billón de años26,27. Su número, tamaño y forma pueden variar según el tipo de célula, tejido u órgano. Poseen dos tipos de ácido desoxirribonucleico (ADN): uno nuclear, que codifica la mayoría de las proteínas necesarias para los procesos metabólicos propios de la mitocondria, y otro independiente del genoma, denominado ADN mitocondrial (ADNmt), que contiene un total de 37 genes que codifican para el ácido ribonucleico (ARN) de transferencia (22 tARN), ARN ribosómicos (2 rARN) y ARN mensajeros (13 proteínas componentes del sistema de fosforilación oxidativa). Esta última característica es única y diferencia a la mitocondria de cualquier otro tipo de organelo. Además, el ADNmt contiene dinucleótidos CpG (islas CpG) hipometilados que se asemejan al CpG del ADN bacteriano y son fundamentales en la activación de las vías de señalización y propagación de la inflamación28.
La estructura de la mitocondria está formada por dos membranas: una externa, lisa y que pertenece a la célula, y otra interna, que se encuentra plegada formando crestas mitocondriales, lo que le permite incrementar su superficie (estimada en 14,000 m2 de membrana interna en el ser humano)27. Esta capa interna pertenece al organelo y es impermeable (carece de poros). En ella se ubican las proteínas de la cadena transportadora de electrones (CTE), las proteínas transportadoras ubiquinona (coenzima Q) y citocromo C (cit C), y los oxisomas o partículas F (complejo enzimático adenosín trifosfato [ATP] sintasa). A su vez, la presencia de una doble membrana permite definir dos espacios, el espacio intermembrana y la matriz mitocondrial, rodeada por la membrana interna, que contiene, entre otras enzimas, las del ciclo de Krebs, el ADNmt y los ribosomas.
La función principal de las mitocondrias es la respiración celular, cuyo fin es producir energía. En la glucólisis, la rotura de una molécula de glucosa origina dos moléculas de ácido pirúvico, que se convierten en acetil-CoA por oxidación y descarboxilación (a través del sistema enzimático piruvato-deshidrogenasa). A su vez, esta se convierte en el principal precursor del ciclo de Krebs. La acetil-CoA dona electrones a la CTE, principalmente al complejo I.
La CTE consiste en un grupo de proteínas y moléculas organizadas en cuatro grandes complejos (I-IV), que se reducen y oxidan al transferirse entre ellas electrones procedentes de las formas reducidas de la nicotinamida adenina dinucleótido (NADH) y del flavín adenín dinucleótido (FADH2), ambas moléculas originadas en las fases más tempranas de la respiración celular.
Como resultado, la energía liberada en la oxidación de estas moléculas, al ser mayor que la consumida en la reducción, se utiliza para bombear protones desde los complejos I, III y IV al espacio intermembrana, generando así un gradiente electroquímico (potencial de membrana de aproximadamente −180 mV). El complejo IV, o citocromo C oxidasa, capta los electrones y los transfiere al oxígeno molecular, el cual es su aceptor final. Esta enzima es la encargada de la mayor parte del consumo de oxígeno del organismo (respiración).
La energía almacenada en este gradiente electroquímico, denominada fuerza protón-motriz, se debe a la diferencia de concentración de protones entre la matriz mitocondrial y el espacio intermembrana, denominada quimiosmosis, permitiendo la traslocación de protones desde el espacio intermembrana hacia la matriz a través de la proteína transmembrana ATP sintasa. La ATP sintasa cataliza la adición de un fosfato al adenosín difosfato (ADP) para finalmente sintetizar ATP. En conjunto, tanto el transporte de electrones como la quimiosmosis constituyen la fosforilación oxidativa (Figura 1).
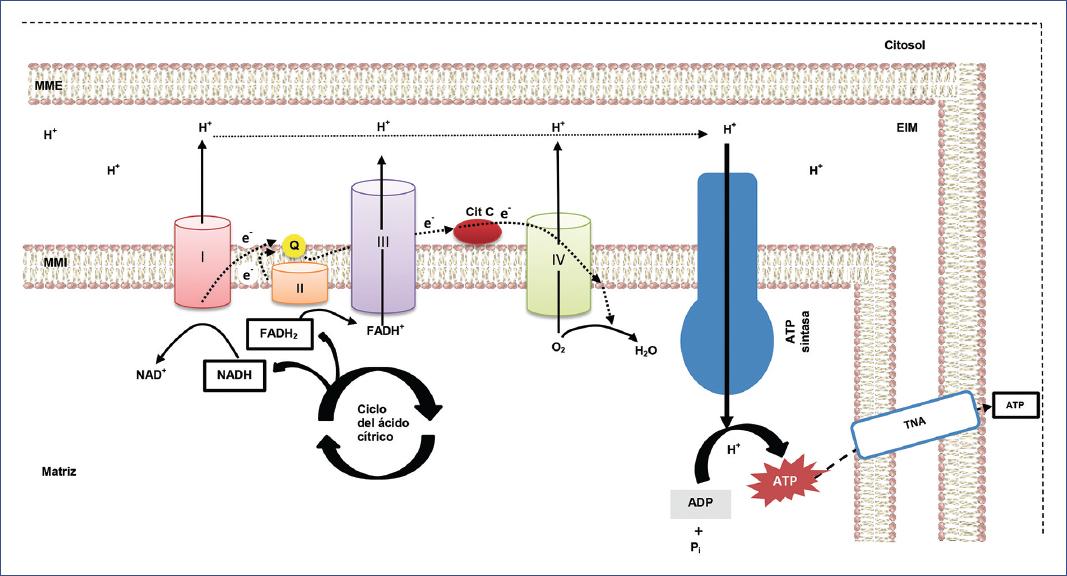
Figura 1 Representación esquemática de la fosforilación oxidativa y su interacción con el ciclo de Krebs. La cadena transportadora de electrones y la ATP sintasa se localizan insertadas en la membrana interna de la mitocondria. Los números romanos indican cada uno de los complejos de la cadena respiratoria. ADP: adenosín difosfato; ATP: adenosín trifosfato; Cit C: citocromo C; FADH+: flavín adenín dinucleótido forma oxidada; FADH2: flavín adenín dinucleótido forma reducida; H+: protón; H2O: agua; MME: membrana mitocondrial externa; MMI: membrana mitocondrial interna; NAD+: nicotinamida adenina dinucleótido forma oxidada; NADH+: nicotinamida adenina dinucleótido forma reducida; O2: oxígeno; Pi: fosfato inorgánico; Q: coenzima Q; TNA: translocador de nucleótidos de adenina.
En condiciones aeróbicas, la eficiente producción energética durante la fosforilación oxidativa representa un riesgo para la célula a consecuencia de la capacidad oxidante del oxígeno y de la formación de pequeñas cantidades de radicales libres (secundaria a la inevitable fuga de electrones) que dañan a las biomoléculas, ocasionando un efecto nocivo en la función y la sobrevida celular29. No obstante, en el paciente crítico con sepsis se ha descrito una importante reprogramación transcriptómica de genes mitocondriales30 y la activación de diversos mecanismos homeostáticos redox con el objetivo final de lograr una «autoprotección celular»31-33.
Homeostasis mitocondrial en el sujeto sano
Con excepción de los glóbulos rojos, todas las células del cuerpo poseen mitocondrias que desempeñan un papel clave en el metabolismo celular. Estas participan en más del 90% de la producción energética mediante la fosforilación oxidativa34, que es un proceso por el cual las enzimas de la CTE transforman el potencial eléctrico transmembrana (ΔΨm) en energía bioquímica. Junto con la fosforilación oxidativa basal, existe una capacidad respiratoria disponible que es la reserva de fosforilación mitocondrial destinada a responder al incremento de las demandas metabólicas (índice de reserva bioenergética).
La función mitocondrial varía en respuesta a factores intracelulares y extracelulares que regulan la homeostasis bioenergética celular. En condiciones de normalidad, el consumo de oxígeno a través de la CTE se encuentra estrechamente ligado a la producción de ATP (sustrato energético de los procesos metabólicos celulares) y regulado por la demanda metabólica (respiración acoplada).
Por otra parte, el oxígeno no empleado en la fosforilación oxidativa (1% del consumo de oxígeno mitocondrial) se destina a la producción de especies reactivas de oxígeno. Esta producción está estrictamente controlada por varias enzimas antioxidantes, como la superóxido dismutasa manganeso o la glutatión oxidasa. Estas enzimas pueden desempeñar una función protectora o nociva en la señalización mitocondrial, dependiendo de la magnitud y la duración de su producción35,36.
Finalmente, la homeostasis mitocondrial requiere un perfecto equilibrio entre la mitofagia y la biogénesis (incremento de la masa mitocondrial celular).
Mortalidad por choque séptico en pediatría
A pesar del conocimiento adquirido en las últimas décadas, la mortalidad hospitalaria de los niños con sepsis se mantiene elevada: del 19% en los países desarrollados y del 32% en aquellos en vías de desarrollo37. Las razones por las cuales los niños fallecen son variables y, por lo tanto, los mecanismos por los que se puede esperar que una determinada intervención afecte la mortalidad requieren especial consideración. A modo de ejemplo, la mortalidad temprana debería ser menos frecuente debido a la precocidad en el reconocimiento y el inicio de la terapia de reanimación en el paciente séptico38. En contraparte, la mortalidad tardía (> 3 días) es atribuible, principalmente, a la persistencia del SDMO39. Así, las nuevas terapias dirigidas a la disfunción orgánica deberían reducir la mortalidad en aquellos niños que logran una estabilización inicial, pero que posteriormente no mejoran.
Síndrome de distrés microcirculatorio y mitocondrial
El paciente con sepsis que presenta una pobre respuesta terapéutica habitualmente tiene unas variables macrocirculatorias relativamente normales, pero con signos microcirculatorios asociados a un mal pronóstico, condición que se ha denominado síndrome de distrés microcirculatorio y mitocondrial (SDMM)40,41. Lo fundamental de esta propuesta es la presencia de hipoxia tisular que persiste tras la normalización de las variables de la macrocirculación42.
La hipoxia citopática se refiere a la alteración en la producción de ATP a pesar de la existencia de unos valores normales o supranormales de oxígeno tisular43,44. Esta condición puede deberse a factores tales como la disminución de la entrega de sustratos clave o la inhibición de etapas dentro del ciclo de Krebs, la alteración de las enzimas de la CTE o el desacoplamiento de la fosforilación oxidativa resultando en la producción de calor más que en la formación de ATP16,45.
Dos mecanismos se han propuesto para explicar el desarrollo del SDMM. El primero es una alteración de la microcirculación que se caracteriza por la disminución de la densidad de los capilares funcionales. De este modo, se incrementan tanto la distancia de difusión del oxígeno46 como la heterogeneidad capilar, generando un shunt microcirculatorio47, lo que implica una reprogramación metabólica: el cambio en la generación de ATP desde la fosforilación oxidativa a la glucólisis aerobia (efecto Warburg) y la inhibición de la CTE48; este segundo mecanismo es el que se ha planteado para el desarrollo del SDMM.
Dicha propuesta conceptual permitiría la identificación específica del compartimento donde ocurre la falla, y posibilitaría la instauración de estrategias terapéuticas adecuadas; las actuales están dirigidas casi en forma exclusiva a la corrección de la macrohemodinamia. Por consiguiente, el choque séptico, en parte, podría describirse fisiopatológicamente como un SDMM.
Respuesta inmunitaria celular y respiración mitocondrial
La respuesta protectora celular se desencadena por la activación de señales de «peligro» ante diferentes estímulos49. Existe evidencia de que los patrones moleculares asociados a patógenos microbianos (PAMP, pathogen-associated molecular patterns) y los patrones moleculares asociados al daño (DAMP, damage-associated molecular patterns), al ser identificados por los receptores de reconocimiento de patrones (PRR, pattern-recognition receptors), como los tipo Toll (TLR, Toll-like receptors), presentes en las células que participan en el sistema inmunitario innato, inician una respuesta sistémica por medio de las vías MyD88/TRADD/NF-κB y JAK1/STAT3 (Janus kinase 1-signal transducer and activator of transcription 3)50. La activación de los TLR facilita la activación de la vía dependiente y de la vía independiente de MyD88. En la vía independiente (MyD88/TRADD/NF-κB) se forma un complejo de señalización con TRADD (TNF-receptor asocciated via death domain) y otras proteínas acopladoras que conduce a la producción del interferón tipo I (IFN-I) y a la expresión de genes inducibles por IFN, y también involucra una fase tardía de activación de NF-κB51. De igual manera, diferentes citocinas utilizan la vía de señalización de JAK1/STAT3 para la transducción de señales desde la membrana celular al núcleo. Una vez que la citocina se une a su receptor, la JAK se activa, lo que estimula al factor de transcripción STAT, que a su vez induce a nivel nuclear los genes implicados en la producción de citocinas, las cuales son indispensables para la inmunidad innata y adaptativa52,53. Probablemente esta respuesta celular es modulada por la mitocondria54.
La activación de las células inmunitarias vía TLR aumenta la transcripción de citocinas proinflamatorias y antiinflamatorias, del factor de necrosis tumoral (TNF, tumor necrosis factor), de interleucinas (IL), de especies reactivas de oxígeno (como peróxido de hidrógeno o radical hidroxilo) y de especies reactivas de nitrógeno, incrementando el estrés oxidativo y nitrosativo. En este último ocurre un aumento de la producción de óxido nítrico que determina su reacción con el anión superóxido, originando peroxinitrito, especie altamente reactiva capaz de oxidar y nitrar componentes celulares y tisulares55. Este mecanismo, junto con el estrés oxidativo, ocasiona daño proteico y del ADN, como el bloqueo de la respiración mitocondrial, originando la disfunción mitocondrial inducida por sepsis (Figura 2)42,56,57.

Figura 2 Sucesión de eventos causantes de disfunción mitocondrial en el paciente con sepsis. Los números romanos indican los complejos de la cadena respiratoria (I-IV). 1) Aumento de la actividad de la enzima óxido nítrico sintasa inducible. 2) Incremento de la producción de anión superóxido mitocondrial. 3) Producción de peroxinitrito. 4) Nitrosilación del complejo respiratorio. 5) Disminución del potencial de membrana. 6) Apertura de poro de transición mitocondrial. La disfunción de la cadena transportadora de electrones (CTE) da como resultado una producción intramitocondrial extrema de especies reactivas de oxígeno (ERO), lo que puede conducir a un daño oxidativo en la membrana, en la actividad de la CTE y el ADN mitocondrial. El incremento de permeabilidad de la membrana mitocondrial produce la liberación de citocromo C en el citosol; lo que conduce a apoptosis. El aumento de la permeabilidad de la membrana también hace que exista reflujo de Ca++ hacia el citoplasma. Las ERO mitocondriales también pueden transportarse al citoplasma e inducir estrés oxidativo, seguido de la activación de vías de señalización de estrés oxidativo que modulan diversas funciones celulares. Finalmente, las ERO liberadas en el espacio extracelular dañarán a otras células y órganos (DAMP, damage-associated molecular patterns). ADNmt: ácido desoxirribonucleico mitocondrial; ADP: adenosín difosfato; ATP: adenosín trifosfato; Ca++: calcio ionizado; e−: electrón; ERO: especies reactivas de oxígeno; FAD+: flavín adenín dinucleótido forma oxidada; FADH2: flavín adenín dinucleótido forma reducida; H+: protón; H2O: agua; iNOS: óxido nítrico sintasa inducible; MME: membrana mitocondrial externa; MMI: membrana mitocondrial interna; NAD+: nicotinamida adenina dinucleótido forma oxidada; NADH+: nicotinamida adenina dinucleótido forma reducida; NO: óxido nítrico; O2: oxígeno; O2−: anión superóxido; ONOO−: peroxinitrito; Pi: fosfato inorgánico; PTPm: poro de transición de permeabilidad mitocondrial; Δψm: potencial eléctrico transmembrana.
Por otra parte, los tratamientos empleados sistemáticamente en el paciente séptico, como antibióticos bactericidas58 y catecolaminas59, también pueden inhibir la respiración mitocondrial. Las catecolaminas, además de su acción hemodinámica, presentan propiedades que afectan a la inmunidad y al metabolismo del paciente grave60. En modelos animales clínicamente relevantes se ha observado una alteración de la respiración mitocondrial relacionada directamente con la dosis requerida de epinefrina61,62. A futuro, esta observación permitiría otra perspectiva sobre su utilización (descatecolaminización)59,63.
Propagación de la inflamación y mitocondrias
Las mitocondrias desempeñan un papel trascendental tanto en la propagación sistémica de la inflamación como en la disfunción de órganos distantes. El ADNmt es particularmente vulnerable al daño ocasionado por el estrés oxidativo y los mediadores proinflamatorios, dada su proximidad con la CTE, la carencia de histonas protectoras, la limitada eficiencia de sus mecanismos de reparación y contener exclusivamente regiones codificadoras64,65. De este modo, el ADNmt que se ha fragmentado es transportado a la matriz mitocondrial o al espacio extracelular (ADNmt extracelular), activando diversas vías inflamatorias debido a su similitud con el ADN bacteriano66-69.
Una vez que el ADNmt ha alcanzado el citosol, se promueve la formación del inflamasoma NLRP3 (nod-like receptor-P3 inflammasome), un complejo multimérico intracelular que desencadena la activación de caspasas inflamatorias, que a su vez generan la maduración proteolítica de la IL-1β y la IL-18, además de promover la expresión de IL-6 y TNF-α, ambos procesos clave en la respuesta inmunitaria innata70.
En la circulación, el ADNmt es reconocido como un potente DAMP71, desencadenando una respuesta inflamatoria sistémica (Figura 3)67,72,73. Los estudios clínicos han demostrado la existencia de una asociación estadística entre sus niveles circulantes y la mortalidad, aunque la mayoría de los reportes son de series relativamente pequeñas y carecen de un protocolo estandarizado para su medición, aspectos que deben considerarse antes de establecer su real utilidad clínica como posible biomarcador74,75. En fecha más reciente se ha evaluado su valor pronóstico76 y también su correlación con el desarrollo de SDMO en niños con sepsis77.
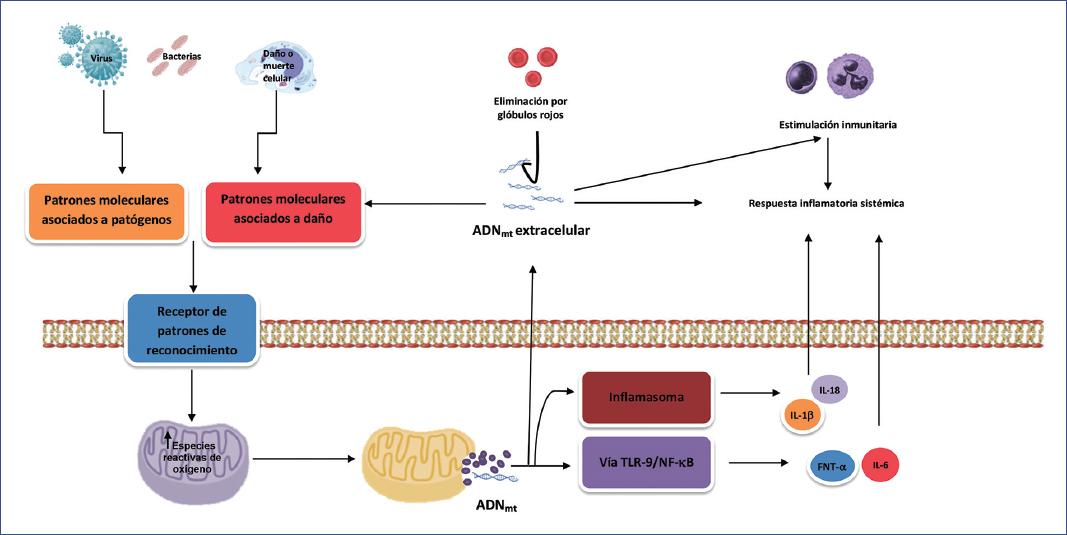
Figura 3 Representación de la producción y liberación del ácido desoxirribonucleico mitocondrial (ADNmt) extracelular. Los patrones moleculares asociados a patógenos (PAMP, pathogen-associated molecular patterns) y los patrones moleculares asociados al daño (DAMP, damage-associated molecular patterns), los cuales han sido liberados ante una infección o daño celular, respectivamente, ocasionan la estimulación de receptores de reconocimiento de patrones (PRR, pattern recognition receptors). Posteriormente se origina la producción de especies reactivas de oxígeno (ERO) mitocondrial, que dañan y fragmentan el ADNmt permitiendo su descompartimentalización y llegada al citosol. Este ADNmt actúa como un potente DAMP, estimulando el inflamasoma o la vía dependiente de los receptores Toll (TLR, Toll-like receptor), TLR-9/factor nuclear-κβ, ocasionando la producción de citocinas proinflamatorias. El ADNmt puede alcanzar el medio extracelular, donde propaga la respuesta inflamatoria inicial al ser reconocido como DAMP por células inmunitarias y no inmunitarias. Se representa la eliminación del ADNmt circulante (cell-free ADNmt) por los glóbulos rojos (vía TLR-9). IL-6: interleucina 6; IL-18: interleucina 8; IL-1β:αinterleucina 1β; FNT-α:αfactor de necrosis tumoral-α.(Modificada de Harrington, et al.75.)
Disfunción mitocondrial en el paciente con sepsis
La relación entre la función mitocondrial y el SDMO aún no se ha esclarecido por completo; sin embargo, existe conocimiento de diversos papeles de las mitocondrias durante la sepsis (Tabla 1).
Tabla 1 Funciones mitocondriales en el sujeto sano y durante el desarrollo de sepsis
| Metabolismo y señalización celular | |
|---|---|
| Fosforilación oxidativa | Fuente primaria de VO2 y VCO2
Prioridades en el uso de energía: transporte de Na+ y Ca+2, síntesis proteica, replicación de ADN y ARN |
| Homeostasis Ca+2 intracelular Generación de especies reactivas de oxígeno y nitrógeno | Esencial para el normal funcionamiento celular
Riesgo potencial ante su exceso |
| Daño mitocondrial y reparación | |
| Apoptosis vía intrínseca: citocromo C | Requiere energía, efecto antiinflamatorio |
| Necrosis por rotura de membrana | Liberación de ADNmt (efecto proinflamatorio,
DAMP) Ocasiona mecanismo de mitofagia |
| Fusión, fisión y biogénesis mitocondrial | Con el objetivo de mantener la salud mitocondrial |
ADN: ácido desoxirribonucleico; ADNmt: ácido desoxirribonucleico mitocondrial; ARN: ácido ribonucleico; DAMP: damage-associated molecular patterns (patrones moleculares asociados al daño); VCO2: producción de dióxido de carbono;
VO2: consumo de oxígeno.
Las células del paciente con SDMO son incapaces de consumir el oxígeno disponible, ocasionando un daño en la CTE que genera un «apagón mitocondrial» y, como consecuencia, se produce deterioro de la bioenergética celular, exacerbación del estrés oxidativo y nitrosativo, incremento de la apoptosis y alteración de vías metabólicas esenciales78-81. Estos hallazgos han sido confirmados tanto en modelos animales21,82 como en humanos83, lo cual ratifica el papel de la disfunción mitocondrial en la patogenia del SDMO54. Brealey, et al.21 comunicaron menores cantidades de ATP en las biopsias muscular de pacientes fallecidos en comparación con los sobrevivientes. En la misma línea, la demostración de una mayor concentración tisular de oxígeno apoya el papel de la hipoxia citopática en la falla orgánica en el choque séptico16.
Finalmente, la restauración espontánea o farmacológica de la disfunción mitocondrial se asocia con recuperación del SDMO y con una mayor sobrevida. En concreto, en animales y en humanos22 se ha evidenciado que se logra la recuperación de la función orgánica y una mayor sobrevida54 cuando mejoran tanto la biogénesis mitocondrial como la mitofagia84-87. No obstante, esto podría tratarse solo de un epifenómeno, pues faltan estudios intervencionales que lo demuestren fehacientemente.
Hibernación celular como causa de disfunción orgánica múltiple
El suministro insuficiente de oxígeno ocasiona hipoxia tisular, mientras que la utilización alterada de este lleva a disoxia. Ambos mecanismos generan una reducción en la producción de ATP, provocando no solo disfunción celular en órganos específicos, sino también pérdida de la integridad celular, ya que mantener su estructura depende de la energía. Así, se podría suponer que, debido a las importantes alteraciones bioquímicas y metabólicas existentes en los pacientes que fallecen, la falla orgánica es consecuencia de una extensa muerte celular88. Sin embargo, los estudios post mortem han revelado una discordancia entre los hallazgos histológicos y la magnitud de la disfunción orgánica en los pacientes sépticos89,90.
Lo previamente mencionado, en concomitancia con un flujo sanguíneo preservado y la existencia de una tensión tisular de oxígeno adecuada91 asociada a un bajo consumo de este, han llevado a cuestionar el papel de la hipoxia tisular como principal mecanismo fisiopatológico en el paciente con SDMO.
Por ello, se necesita un paradigma que pueda explicar la existencia de la disfunción orgánica en ausencia de un daño estructural significativo y, aún más, ante un aporte adecuado de oxígeno92.
Una consecuencia notable de la activación de las vías de respuesta celular ante la presencia de señales de peligro es la supresión de las actividades dependientes de energía en favor de aquellas que son esenciales para la sobrevida celular, lo que se ha corroborado en cardiomiocitos (hibernación miocárdica)93,94, hepatocitos95 y neumocitos (conformidad hipóxica)96.
En esta misma línea, quizás el fenómeno de parálisis inmunitaria (vide infra), descrito en el paciente séptico97, refleje una hibernación leucocitaria en vista de que la respiración mitocondrial de las células mononucleares periféricas circulantes no puede responder ante un incremento de la demanda metabólica98-101. Asimismo, la escasa cantidad de células epiteliales tubulares necróticas observadas en el paciente con falla renal aguda es consistente con el concepto de hibernación epitelial renal102. Esta idea no es nueva, pues hace más de cuatro décadas ya se señaló que la falla renal aguda era, más bien, un «éxito» renal agudo, pues esta respuesta adaptativa permitía al riñón «ahorrar» en tareas altamente dependientes de energía (reabsorción tubular), configurando así un mecanismo de protección103.
El paradigma de la inflamación exacerbada no logra explicar por completo los eventos observados en los pacientes con sepsis. De hecho, durante este proceso también se liberan citocinas antiinflamatorias que buscan regular la respuesta inmunitaria, llegando a desarrollar, en ocasiones, un síndrome de respuesta antiinflamatoria compensatoria (CARS, compensatory anti-inflammatory response syndrome). La inmunoparálisis o CARS consiste en una hiperactividad de esta respuesta antiinflamatoria caracterizada por la alteración en la expresión del HLA-DR monocitario (mHLA-DR, monocytic human leukocyte antigen-DR), apoptosis linfocitaria y aumento de citocinas reguladoras104, mecanismos que parecen influir en el desarrollo de infecciones secundarias y la muerte del paciente105.
Lo previamente señalado sugeriría que el SDMO es un fenómeno más bien de carácter funcional que estructural, basado en una reducción primaria temporal del metabolismo celular79,106. Así, la falla multiorgánica puede verse como una respuesta adaptativa y protectora que ayudaría a prevenir la muerte celular (Figura 4).

Figura 4 Mecanismos involucrados en la disfunción metabólica y apagón mitocondrial. ATP: adenosín trifosfato; CTE: cadena transportadora de electrones; ERO: especies reactivas de oxígeno; ERN: especies reactivas de nitrógeno; DAMP: damage-associated molecular patterns; FO: fosforilación oxidativa; O2: oxígeno.
Carcillo, et al.107 han señalado que el término «disfunción mitocondrial» sería una denominación errónea, ya que la regulación a la baja o la reducción temporal de la actividad metabólica observada en modelos experimentales probablemente representa una respuesta adaptativa (estado hipometabólico), hallazgo que con frecuencia se observa en los pacientes muy graves80,108.
Al parecer, esta idea estaría avalada por la observación de que la función orgánica usualmente se restaura (en días a semanas) en los pacientes sobrevivientes de SDMO, incluso en órganos con una pobre respuesta regenerativa, lo que indica que la disminución de la actividad mitocondrial es adaptativa e inicialmente reversible49,79,106.
El cambio desde el estado de apagón mitocondrial109 al de activación de la biogénesis22 se encuentra finamente regulado por diversos factores, los cuales dependerán de la gravedad de la sepsis, de factores genéticos110, de las características del individuo (edad, enfermedades asociadas) y de la terapia empleada111.
En suma, el SDMO refleja una respuesta funcional adaptativa, transitoria, protectora y potencialmente reversible, más que una lesión estructural, en el paciente con sepsis42.
Monitorización de la función mitocondrial
En la actualidad, la evaluación de la función mitocondrial se encuentra limitada al ámbito experimental y preclínico, principalmente mediante métodos ex vivo, lo que podría no ser representativo de una situación in vivo. Esto se debe a que la mitocondria es un organelo subcelular, y también a la comprensión inexacta y cabal de los procesos bioquímicos complejos que involucran a las reacciones redox, así como a la incapacidad de realizar mediciones confiables en tiempo real en medios biológicos112.
Se dispone de técnicas in vivo, como la fluorometría para NADH, la espectroscopía por resonancia magnética y la espectroscopía cercana al infrarrojo (NIRS, near infrared spectroscopy) para medir el estado redox de la enzima citocromo C oxidasa113. Por otra parte, la tensión de oxígeno mitocondrial se puede evaluar en la epidermis mediante la fluorescencia de protoporfirina IX114.
Aunque la magnitud de la disfunción mitocondrial inducida por la sepsis es variable en los diversos sistemas orgánicos comprometidos115, existe evidencia experimental en un modelo de choque hemorrágico de que la reducción de la respiración mitocondrial en células mononucleares periféricas se correlaciona con cambios similares en las mitocondrias del riñón y del corazón116. Por otro lado, en células mononucleares periféricas de niños sépticos se correlacionó el ΔΨm durante las primeras 48 horas con la magnitud del daño orgánico a la semana. Se evidenció un mayor ΔΨm en aquellos pacientes con función orgánica normal al séptimo día en comparación con los que mostraron persistencia del SDMO posteriormente83. Weiss, et al.117 demostraron que la permanencia de una respiración mitocondrial disminuida en células mononucleares periféricas se asocia con una lenta recuperación de la función orgánica. Sin embargo, en esta misma línea, se requieren mayores estudios para la obtención de resultados concluyentes.
Terapia farmacológica para la disfunción mitocondrial
El listado de los agentes farmacológicos destinados a prevenir o tratar la disfunción mitocondrial en el paciente con sepsis es extenso (Figura 5). Estos pueden ser clasificados de una manera esquemática en las siguientes categorías: entrega de sustratos, cofactores y donantes de electrones favorecedores de la fosforilación oxidativa; aporte de antioxidantes exógenos y depuradores de radicales libres; estabilizadores de la membrana mitocondrial; y terapia hormonal (Tabla 2)118,119.
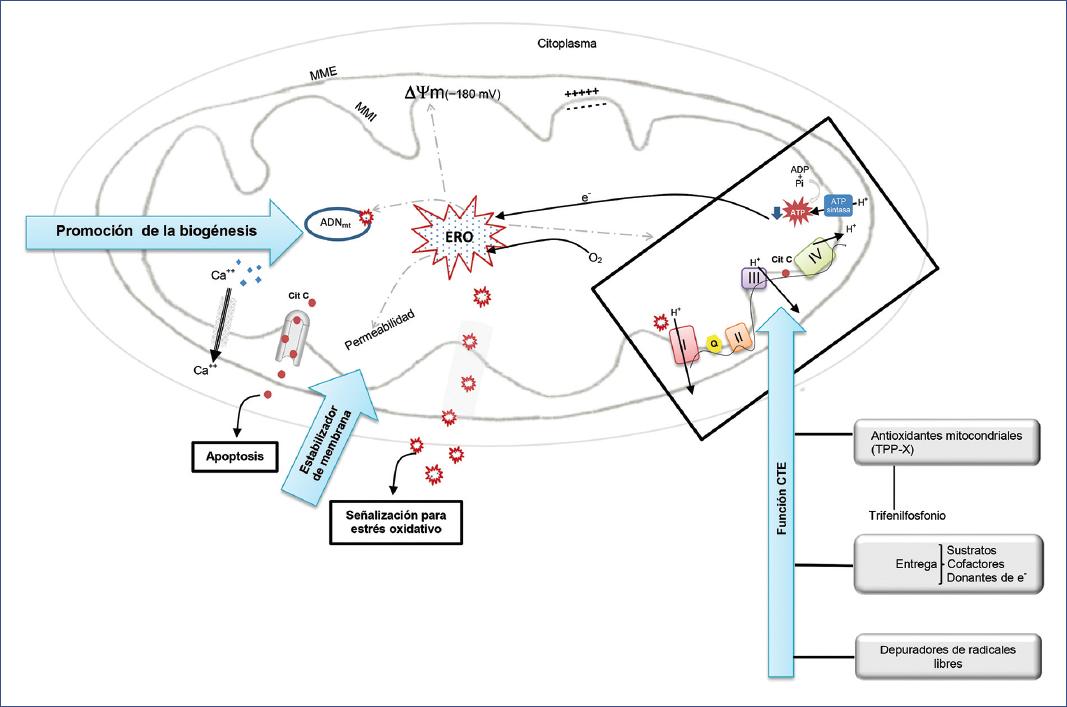
Figura 5 Estrategias terapéuticas con diana en la mitocondria. Los números romanos indican cada complejo de la cadena respiratoria. La terapia más promisoria son los antioxidantes conjugados con cationes (trifenilfosfonio, TPP+), los cuales se acumulan específicamente en las mitocondrias y mejoran la función de la cadena transportadora de electrones. La estabilización de la membrana inhibe que las especies reactivas de oxígeno provoquen más lesiones y protege a las mitocondrias de la inflamación y la rotura, reduciendo así la fuga de moléculas que causan apoptosis y alteración del calcio en el citoplasma. La promoción de la biogénesis mitocondrial reactiva las expresiones del ADNmt, mejorando así la expresión de proteínas mitocondriales. ADNmt: ácido desoxirribonucleico mitocondrial; ADP: adenosín difosfato; ATP: adenosín trifosfato; Ca++: calcio ionizado; Cit C: citocromo C; e−: electrón; ERO: especies reactivas de oxígeno; H+: protón; MME: membrana mitocondrial externa; MMI: membrana mitocondrial interna; O2: oxígeno; Pi: fosfato inorgánico; Q: coenzima Q; ΔΨm: potencial eléctrico transmembrana. (Modificada de Zhang, et al.24.)
Tabla 2 Agentes farmacológicos propuestos para la prevención o la disminución de la disfunción mitocondrial séptica
| Relacionados con la matriz mitocondrial y la cadena transportadora de electrones | Antioxidantes mitocondriales y depuradores de radicales libres | Estabilizadores de la membrana mitocondrial | Terapia hormonal y otros |
|---|---|---|---|
| Succinato | Mitoquinonac, d | CsA/NIM 811 | Glucocorticoides |
| Glutamina | Mito-Vit-Ec, d | Tetrametilpirazina | Insulina |
| ATP-MgCl2 | Péptidos SSc | Metformina | Melatonina |
| L-carnitina | Mito-TEMPOc, d | Imeglimina | Estrógenos |
| Coenzima Q (ubiquinona) | Tetrametilpiperidina (tempol)c | Ciclosporina | Leptina |
| Citocromo C | Inhibidores NOS | CeO2 NPs | |
| Cafeína | N-acetilcisteína | ||
| Ácido tióctico (ácido a-lipoico) | Etilpiruvato | ||
| rhTFAMa | Inductor HO | ||
| H2 Sb | Fenol (resveratrol) | ||
| NOb | GSH | ||
| COb |
aPromotor de la biogénesis.
bGasotransmisor.
cAntioxidante sintético exógeno con diana en la mitocondria.
dCatión lipófilo.
ATP-MgCl2: adenosín trifosfato-cloruro de magnesio; CeO2 NPs: nanopartículas de óxido de cerio; CO: monóxido de carbono; CsA: ciclosporina A; GSH: glutatión; HO: hemo oxigenasa; H2 S: sulfuro de hidrógeno; NIM 811: N-metil-4-isoleucina; NO: óxido nítrico; NOS: óxido nítrico sintetasa; SS: péptidos Szeto-Schiller; rhTFAM: factor A de transcripción mitocondrial recombinante humano.
Además, es relevante conocer el momento adecuado de su aplicación o uso para una correcta evaluación de su eficacia: a) prevención y restitución precoz de la disfunción mitocondrial; b) una vez establecida, prevención del colapso energético celular; c) en el periodo de biogénesis mitocondrial; y d) en el periodo de reparación o sustitución de aquellas mitocondrias que se encuentran dañadas.
Experimentalmente, estas terapias han demostrado una disminución del estrés oxidativo y de las citocinas inflamatorias circulantes, junto con una restauración de la generación de ATP. Se han reportado los hallazgos de estudios preclínicos en modelos animales que han evaluado el efecto de diversos tipos de agentes sobre la función orgánica (Tabla 3)120-124. No obstante, aún queda pendiente la valoración de su eficacia clínica125-127.
Tabla 3 Trabajos experimentales en animales que evalúan el uso de diversas terapias farmacológicas antioxidantes mitocondriales y no antioxidantes
| Autor, añoref | Sepsis | Animal/modelo | Agente utilizado | Efectos orgánicos | Efectos clínicos observados | Mortalidad |
|---|---|---|---|---|---|---|
| Lowes, et al., 2008120 | Sí | Ratones LPS-PG |
Antioxidante mitocondrial (MitoQ) |
Disminución de marcadores bioquímicos de disfunción hepática Disminución de marcadores bioquímicos de disfunción renal | — | — |
| Patil, et al., 2014121 | Sí | Ratones LCP |
Antioxidante mitocondrial (Mito-TEMPO) |
Mejoría de la microcirculación renal Mejoría de la tasa de filtración glomerular | — | Incremento a las 96 h en la sobrevida del 40% al 80% |
| Selvaraj, et al., 2015122 | Sí | Ratones LPS |
No antioxidante (CeO2 NP) |
Disminución del daño hepático | Normalización de temperatura, frecuencia respiratoria y presión arterial | Disminución de la mortalidad del 70% al 10% |
| Xu, et al., 2020123 | Sí | Ratones LPS |
Mixto (estrógeno/Mito-TEMPO) | Mitigación del daño hepático con disminución de AST y
ALT Menor grado de infiltración hepática por células inflamatorias |
— | — |
| Xu, et al., 2020124 | Sí | Ratones LCP |
Nutrientes-antioxidantes (ascorbato, taurina, glutatión) |
Mitigación del daño hepático con disminución de ALT
Menor grado de infiltración hepática por células inflamatorias Menor grado de necrosis hepatocelular Disminución de creatinina plasmática |
— | — |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CeO2 NP: nanopartículas de óxido de cerio; LCP: ligazón cecal y punción; LPS: lipopolisacárido;
PG: peptidoglicano
A futuro
Un desafío en el manejo de los pacientes con sepsis muy graves es reconocer el momento en que los esfuerzos terapéuticos, muchos de ellos orientados a la búsqueda de la normalidad fisiológica, pueden inducir modificaciones dañinas para la adecuada puesta en marcha del intento alostático adaptativo128,129.
La pronta identificación y el conocimiento del subgrupo de pacientes sépticos que se beneficiarían de una resucitación metabólica es esperable que se obtengan a través de la medicina de precisión mediante transcriptómica, metabolómica y farmacogenómica31,130. Como la mayoría de las intervenciones terapéuticas en el paciente séptico, el momento en que estas se efectúen es de suma relevancia, ya que el organismo puede defenderse ante el incremento prematuro de su metabolismo.
La capacidad de identificar y seguir los cambios de la función mitocondrial (evaluación bioenergética) en el niño en riesgo de prolongación de la disfunción orgánica y que podría beneficiarse de una terapia mitocondrial será, entonces, un paso clave.
Finalmente, una atractiva e interesante área de desarrollo es la inducción farmacológica de un estado hipometabólico bajo demanda, ya sea global u orgánico, proceso denominado «animación suspendida»131,132.
El papel de la hipoxia citopática y la reanimación metabólica son campos de investigación activa. No obstante, los mecanismos precisos que están involucrados en la falla multiorgánica permanecen desconocidos.
Existe suficiente certeza para apoyar que la disfunción mitocondrial es clave en la fisiopatología del SDMO y que se caracteriza por una producción reducida de ATP con un incremento del estrés oxidativo. La evidencia orienta a la existencia de un apagón metabólico adaptativo originado por una reducción del metabolismo celular, en particular de la fosforilación oxidativa, priorizando la utilización de la energía para mantener la homeostasis del ATP.
El ADNmt liberado es reconocido como un importante desencadenante de la inflamación sistémica, que daña múltiples órganos y se asocia a mortalidad en los pacientes gravemente enfermos.
Existe controversia al analizar la disfunción mitocondrial en enfermedades graves, especialmente en modelos animales de sepsis, ya que no siempre se consideran las diferencias entre especies y entre órganos, o el momento en que se realiza su evaluación. Por esto, es necesario buscar modelos más representativos y que permitan su extrapolación al entorno clínico.
Los agentes farmacológicos para la prevención y el tratamiento de la disfunción mitocondrial, como aquellos para la inducción terapéutica de la biogénesis, son opciones atractivas y se constituyen en una promisoria línea de investigación para la resucitación metabólica. Sin embargo, ninguna se ha reflejado en la práctica clínica actual.
La monitorización de la oxigenación tisular y la función mitocondrial confiere un periodo ventana para determinar la suficiencia de la perfusión orgánica y del bienestar celular en el paciente séptico muy grave. Por lo tanto, queda pendiente definir quiénes son los pacientes candidatos a recibir terapia orientada a la mitocondria.

