











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Purine metabolism constitutes a key pathway for every organism. It is involved in DNA and RNA synthesis and degradation, and involves the production of ubiquitous metabolites, such as adenosine triphosphate (ATP) (Yin et al., 2018). There are two main routes for the synthesis of nucleotides, the de novo biosynthetic pathway and the complementary salvage pathway. The de novo biosynthetic pathway is responsible for the production of inosine monophosphate (IMP) in a twelve-step biosynthetic process with consumption of ATP (Pang et al., 2012; Yin et al., 2018). The complementary salvage pathway, however, converts extra-cellular nucleobases or degrades purine metabolites into the corresponding nucleosides and nucleotides (Nelson et al., 2008), and obviates energetically expensive de novo synthesis of these compounds.
When mammals are exposed to oxygen limiting situations, such as breath-hold diving induced hypoxia or ischemia, systemic and intracellular changes operate together to minimize injury and restore adequate oxygenation. During hypoxia, mammalian tissues cannot produce enough energy to maintain essential cellular processes (Giordano, 2005); thus, a reduction of ATP synthesis is produced, promoting the degradation of purine nucleotides (Nelson et al., 2008) which are accumulated in the cells (Raivio et al., 2001). The catabolic compounds derived from ATP breakdown, hypoxanthine (HX), inosine (I), xanthine (X), uric acid (UA) and adenosine (A), have been widely used as markers of tissue hypoxia (Maiuolo et al., 2016).
Purine degradation involves the activation of purine nucleotide phosphorylase (PNP, E.C. 2.4.2.1) and xanthine oxidase (XO, E.C. 1.1.3.22) (Maiuolo et al., 2016). PNP catalyzes the reversible conversion of nucleosides (guanosine, xanthosine, inosine) to purine bases (guanine, xanthine, and hypoxanthine) (Dudzinska et al., 2006), while XO catalyzes the oxidation of hypoxanthine and xanthine to uric acid (Vorbach et al., 2003). Salvage of purines occurs through two main enzymes, adenine phosphoribosyltransferase (APRT), which mediates the transfer of phosphoribosyl-1-pyrophosphate (PRPP), a high energy sugar phosphate, to adenine, to form adenosine monophosphate (AMP), and hypoxanthine-guanine phosphoribosyltransferase (HGPRT, E.C. 2.4.2.8), which forms IMP and guanosine monophosphate (GMP) (Nelson et al., 2008; Zhang et al., 2008). The enzyme inosine 5-monophosphate dehydrogenase (IMPDH, E.C. 1.1.1.205) uses IMP to produce xanthosine 5-monophosphate (XMP) and, ultimately, ATP (Cherin et al., 2006).
Oxygen limitation has a tissue-dependent effect (Wagner, 2008), which triggers transcriptional (Huang et al., 2004; Rocha, 2007) and posttranslational (Schumacker, 2011) modifications to the enzymes involved in the nucleotide synthesis and degradation. The de novo biosynthetic pathway is energy demanding, requiring five ATP molecules, two glutamine and formate molecules, and one glycine, aspartate and carbon dioxide molecules to generate one IMP molecule. During 24h hypoxia in human cells, a significant increase in hypoxanthine (HX) level is produced (Nagao et al., 2018), which is used by XO. Hypoxia, as well as other stimuli, including growth factors, cytokines and hormones, upregulates XO mRNA expression (Battelli et al., 2016). However, XO activity increases in response to hypoxia without changes in mRNA or protein levels (Berry and Hare, 2004), indicating that XO is modulated at a posttranslational level, mainly through phosphorylation (Kayyali et al., 2001).
Purine recycling in mammals increases in response to prolonged fasting and ischemia (López-Cruz et al., 2014). PNP activity increases significantly during ischemia (Vannoni et al., 2004) and its enzymatic deficiency in purine metabolism leads to several diseases, including Lesch-Nyhan syndrome, clinical gout, nephropathy and neurological disease (Nyhan, 2005). HPGRT activity in mammals plasma indicates an elevated purine recycling rate to avoid accumulation of non-recyclable purines (xanthine and uric acid) (López-Cruz et al., 2014), which rise the need for a comprehensive understanding of the regulation of enzymes involved in the purine metabolism.
The aim of this study was to analyze and compare the enzymatic activity and transcript expression of the proteins involved in purine degradation (XO, PNP) and salvage pathway (HPGRT), and the concentration of metabolites involved in purine metabolism after hypoxic conditions in human muscle cells.
Material and methods
Cell culture
Human skeletal muscle cells (36 cell culture boxes) were cultured in DMEM/F12 50 (Cellgro) supplemented with 15% fetal bovine serum (FBS, Gibco), antibiotics (50 U mL-1 penicillin, 50 µg mL-1 streptomycin), 2 mM glutamax (Gibco), 25 mM HEPES (pH 7.4) and 100 µM sodium pyruvate (Gibco). Cells (18 culture boxes) were cultured in a humid atmosphere at 35°C and serially passaged upon 80% confluence. Hypoxic conditions were obtained by placing the cells (18 cell culture boxes) in a sealed hypoxic workstation for 1 h, medium degas time took around 5 minutes. Afterwards, control cells (normoxia) and hypoxic cells (hypoxia) with 80% confluency were detached from the box using trypsin 0.25% (GIBCO) by a 4 min incubation at 35°C, then trypsin was inactivated by the addition of fresh medium. Cell suspension was recovered in 15 mL tubes, centrifuged at 1200 rpm for 10 min. Supernatant was discarded and the pellet (cells) was re-suspended in 500 µL of medium containing 1% DMSO for metabolite and protein analysis, and in tubes containing TRIzol® Reagent (15596, Invitrogen, Carlsbad, CA, USA) for gene expression. This process was performed for nine cell culture boxes for each treatment. All samples were stored at -80°C until further analysis.
Protein extraction and quantification
Prior to analysis, cells (nine cell culture boxes per treatment) in medium containing DMSO were centrifuged at 1200 rpm for 15 min to remove the medium. Afterwards, 350 µL of deionized water were added and then vortexed for 30 s, followed by three cycles of freezing/thawing for cellular disruption. Finally, samples were centrifuged at 17900 x g for 15 min at 4°C. Supernatant, containing soluble proteins was recovered and stored at -80°C until further analysis. Total soluble protein was quantified using a commercial kit (Bio-Rad Laboratories, Inc. Hercules, CA, USA) based on the method described by Bradford (1976) . Bovine serum albumin (BSA) was used as standard, and results expressed in mg mL-1.
Purine metabolite concentration
Concentration of purines (HX, X, A, I, NAD+, UA, IMP, AMP, ADP, ATP, XMP, GDP and GTP) was quantified in human muscle cell culture samples from normoxia and hypoxia experiments by high performance liquid chromatography (HPLC) (Waters 2695, Milford, MA, USA) (Gianattassio et al., 2003). Samples were treated with 0.5 M cold perchloric acid to precipitate soluble protein, and the solution was then neutralized with 0.5 M potassium hydroxide. The potassium perchlorate produced from the neutralization was eliminated by centrifugation at 17900 x g for 10 min at 4°C. The supernatant containing the purine metabolites was recovered and filtered through a Nylon membrane filter of 0.22 µm (Int. Millipore, Bedford, MA, USA) into a HPLC vial for analysis, the medium was also analyzed to evaluate if the metabolites were expelled from the cell during hypoxia. Purine metabolites were analyzed by HPLC in an analytical column Supelcosil C-18 (150 x 4.6 mm ID, 3 µm particle size; Waters). Metabolites were separated using a gradient from 0 to 19 min, beginning at 100% of buffer A (100 mM KH2PO4, 8 mM TBA pH 6.0) to 50% buffer B (100 mM KH2PO4, 8 mM TBA, 30% acetonitrile pH 6.0) at a flow rate of 1.3 mL min-1. The eluate was monitored at 214 nm. The concentration of each purine metabolite was quantified using standard curves of a purine standards mixture (0.625 to 100 µM), expressing the metabolite concentrations as µM mg-1 protein.
Inosine 5’-monophosphate dehydrogenase (IMPDH, E.C. 1.1.1.205) activity
IMPDH activity was measured by quantifying the concentration of xanthosine 5ˊ-monophosphate (XMP), resulting from the conversion of IMP, using HPLC (Waters 2695, Mildford, MA, USA) (Glander et al., 2001). Enzymatic assays were performed in 40 mM sodium phosphate buffer pH 7.4, containing 0.5 mM IMP, 0.25 mM NAD+ and 50 mM KCl. Reactions were incubated for 3 h at 37°C, subsequently 4 M cold perchloric acid was added to stop the reaction; the reaction mixture was neutralized with 5 M potassium carbonate and finally centrifuged at 15800 x g for 5 min, the supernatant was recovered and filtered through a nylon membrane filter of 0.22 µm (Int. Millipore, Bedford, MA, USA) into an HPLC vial for analysis. XMP was analyzed by HPLC on an analytical column ODS hypersyl 125 AQ (250 x 3.1 mm ID, 3 µm particle size; Waters). XMP was separated using a gradient from 2 to 20 min, beginning at 100% of buffer A (100 mM KH2PO4, 8 mM TBA pH 6.0) to 60% buffer B (100 mM KH2PO4, 8 mM TBA, 30% acetonitrile pH 6.0) at flow rate of 1.1 mL min-1. The eluate was monitored at 214 nm. The XMP concentration was quantified using XMP standard curves (1.5 to 50 µM). One unit of IMPDH is defined as the amount of enzyme needed to produce 1 mM of XMP, and the activity expressed as µM mg-1 protein h-1.
Purine nucleotide phosphorylase (PNP, E.C. 2.4.2.1) activity
PNP activity was measured indirectly by the oxidation of 3, 5-dichloro-2-hydroxybenzenesulfonic acid/4-amino- phenazone to N-(4-antipiril)-3-chloro-5-sulphonate-b-benoquinonamonio, by the hydrogen peroxide formed in the PNP-xanthine oxidase reaction (Chu et al., 1989). Briefly, the enzymatic reaction was performed in 22 mM potassium phosphate buffer pH 7.5, containing 25 U XO, 2000 U of horseradish peroxidase (HRP), 160 mM 4-aminoantipyridine, 120 µM potassium ferrocyanide, 8 mM 3,5-dichloro-2-hydroxybenzenesulfonic acid, and 12 mM inosine. Production of N-(4-antipyryl)-3-chloro-5-sulfonate-p-benzoquinone-monoimine was recorded at 520 nm in a spectrophotometer (Beckman Coulter DU 800, Fullerton, CA, USA) and the change of absorbance was recorded every 5 s for 3 min. The activity of PNP expressed as units (U) mg-1 protein, defining a unit of PNP activity as the amount of enzyme necessary to deplete 1 µM of inosine per min at 25°C.
Hypoxanthine-guanine phosphoribosyltransferase (HG- PRT, E.C. 2.4.2.8) activity
HGPRT activity was measured by quantifying the rate of IMP production, which is oxidized by IMPDH, using the PIERCE HPRT Assay Kit (NOVOCIB, Lyon, France) following the manufacturer’s instructions. Briefly, enzymatic assay was performed in reaction buffer (1X) containing IMPDH, DTT, NAD+ and PRPP. The reaction mixture was recorded every 5 min for 120 min at 340 nm using a microplate reader (Multiskan FC, Thermo Scientific, Finland), and HGPRT activity expressed as nmol mg-1 protein h-1.
Xanthine oxidase (XO, E.C. 1.17.3.2) activity
XO activity was measured using an Amplex® Red Xanthine/Xanthine oxidase assay kit (A22182; Invitrogen, Paisley, UK). The quantification is based on the superoxide anions (O •-) produced by XO, which are degraded to hydrogen peroxide (H O ), which reacts stoichiometrically with Amplex® Red reagent to generate the oxidation product, resofurin. Briefly, the enzymatic assay was performed in reaction buffer (1X) containing 100 µmol L-1 Amplex Red reagent solution, 0.4 U mL-1 HRP and 200 µmol L-1 hypoxanthine, incubated for 30 min at 37°C. The presence of resofurin was recorded at 550 nm using a microplate reader (Multiskan FC, Thermo Scientific, Finland), and the XO activity expressed as mU mg-1. One unit of XO activity is defined as the amount of enzyme required to produce 1 µM of UA from HX per min at 25°C.
Quantification of XO, PNP, IMPDH and HGPRT transcripts RNA extraction and cDNA synthesis
Total RNA was isolated from 1 x 106 cells of skeletal muscle cells from human cell cultures (normoxia and hypoxia) using TRIzol® Reagent (15596, Invitrogen, Carlsbad, CA, USA) according to the manufacturer´s instructions. Contaminating genomic DNA was removed using DNase I (AMPD-1, Sigma-Aldrich Chemical), and total RNA concentration was spectrophotometrically determined at 260 nm. RNAs (1 µg) were reverse transcribed into cDNA using the QuantiTect® Reverse Transcription kit (205313, Qiagen) using random hexamer primers and according to the manufacturer instructions. cDNA samples were stored at -80°C until further analysis.
Real-time PCR analysis
Determination of relative mRNA expression of IMPDH, XO, HGPRT and PNP was performed by real-time PCR, using the ABI 7000 Real Time PCR (Applied Biosystems). cDNA was diluted (1:5) and analyzed using the QuantiTect® SYBR® Green PCR Kit (204145, Qiagen). The reaction mixture contained 10 µL of SYBR® Green, 1.0 µL of forward and reverse primers (10 µM each), 5.0 µL of cDNA (50 ng), and 4.0 µL of sterile, distilled water. Controls without template were included to ensure absence of contaminating DNA. PCR reactions were carried out as follows, an initial cycle at 95°C for 15 min to activate the HotStarTaq® DNA polymerase, followed by the denaturalization of the cDNA at 94°C for 15 s, 40 cycles at 94°C for 15 s each, 60°C for 30 s, and 72°C for 30 s. The specificity of each primer set was validated by the construction of a melting curve, constructed after PCR by increasing the temperature from 75°C to 95°C at a rate of 0.3°C every 20 s. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was determined as housekeeping gene. The relative expression of target mRNA was normalized to the amount of GAPDH in the same cDNA using the standard curve method. Amplification efficiency for each analyzed gene was ~98%, using dilution from 0.25 x 10-2 ng µL-1 to 0.25 x 10-6 ng µL-1 of PCR fragments. Each RT-PCR data point is average of three independent biological replicates. Primer sequences and accession numbers for the housekeeping gene GAPDH and for the different enzyme genes are included in Table 1.
Table 1 Primer sequences for real-time PCR.
Tabla 1. Secuencias de
oligonucleótidos para PCR en tiempo real.
| Target gene | Sense-primer (5´-3´) | Antisense-primer (5´-3´) | GenBank accession no. | Size (bp) |
|---|---|---|---|---|
| IMPDH | GGCTCCATCTGCATCACCCA | CCATGCCCCGGTACTTCTTG | NM_000883 | 262 |
| HPRT | GACTGAAGAGCTACTGTAATG | CACCAGCAAGCTTGCAACC | NM_000194 | 198 |
| PNP | CTGTGTGATGATGCAGGGCAG | TCATTGGGCCCTCTGAGAGG | NM_000270 | 225 |
| XO | ACAGAACACCATGAAGACCC | CTGCCACCAGTTATCAGCATG | NM_000379 | 224 |
| GADPH | GGAAACTGTGGCGTGATGG | CCTGCTTCACCACCTTCTTG | AF261085 | 216 |
Ethical aspects
Muscle samples were obtained in accordance with institutional ethical standards and with the 1964 Helsinki declaration and its later amendments from healthy volunteers subjected to programmed cesarean section at the Hospital de Especialidades Médicas Fidepaz in La Paz, Baja California Sur, México. All participants in the study signed the informed consent form, which, together with the research project, were approved by the Baja California Sur Chapter of the National Mexican Academy of Bioethics and registered with the Comité de Ética en Investigación (CONBIOÉTI- CA-09-CEI-009-20160601).
Statistical analyses
Data were tested for normality and homoscedasticity using Kolmogorov-Smirnov (p < 0.05) and Levene (p < 0.05) tests, respectively. Significant differences in the non-parametric data were determined with Kruskal-Wallis and Mann-Whitney U tests (Zar, 2009). Statistical significance was considered when p ≤ 0.05, determined using GraphPad Prism version 3.03.
Results
Enzyme activity
Figure 1 show the activity of IMPDH, PNP, XO and HGPRT from human skeletal muscle cells after normoxia and hypoxia conditions. IMPDH activity was significantly higher in normoxic (46.59 ± 11.95 µM mg-1 protein h-1) than hypoxic cells (22.09 ± 2.74 µM mg-1 protein h-1) (p < 0.036). There were no significant differences in PNP, XO and HGPRT activities between treatments (p > 0.05).
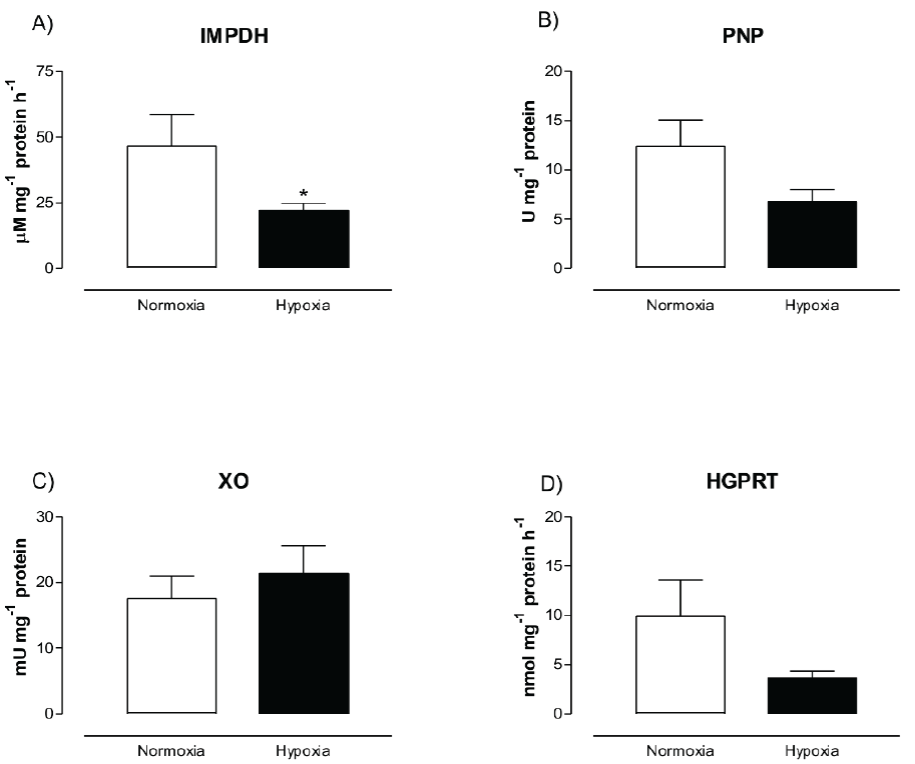
Figure 1 Effect of hypoxia on enzymes involved in purine metabolism. Inosine 5’-monophosphate
dehydrogenase (IMPDH) activity, B) purine nucleotide phosphorylase
(PNP) activity, C) xanthine oxidase (XO) activity and D)
hypoxanthine-guanine phosphoribosyltransferase (HGPRT) activity.
Skeletal muscle cells were incubated under normoxic or hypoxic
conditions for 1 h and cells were collected and tested for each
enzymatic activity. Data are shown as mean ± S.E. of at least five
independent biological experiments by triplicate. * denotes
differences between treatments (p < 0.05).
Figura
1. Efecto de la hypoxia en las enzimas involucradas en el
metabolism de purinas. A) Actividad de inosina monofosfato
dehidrogenasa (IM- PDH), B) Actividad de purina nucleótido
fosforilasa (PNP), C) Actividad de Xantina oxidasa y D) hipoxantina
guanina fosforibosil transferasa (HPGRT). Las células de músculo
esquelético fueron incubadas bajo condiciones de normoxia o hipoxia
por 1 h y las células fueron colectadas y evaluadas para cada ensayo
enzimático. Los datos representan medias ± D.S. de al menos cinco
experimentos biológicos independientes realizados por triplicado.
Asteriscos (*) denotan diferencias estadísticas entre tratamientos
(p < 0.05).
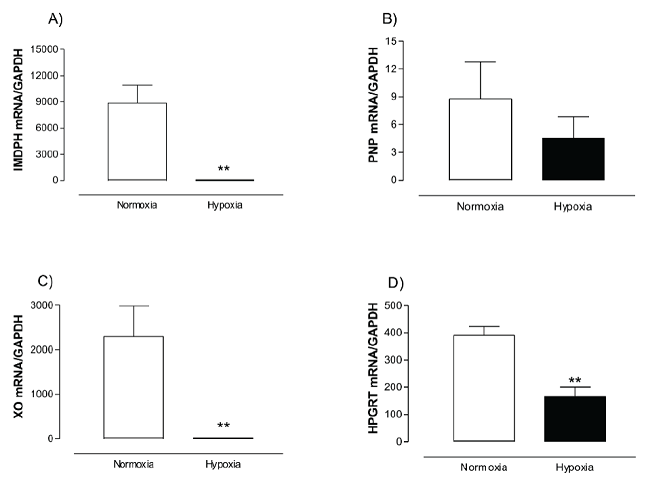
Figure 2 Effect of hypoxia on the expression of the enzymes involved in purine metabolism. A)
Inosine 5’-monophosphate dehydrogenase mRNA (IMPDH), B) purine
nucleotide phosphorylase mRNA (PNP), C) xanthine oxidase mRNA (XO),
and D) hypoxanthine-guanine phosphoribosyltransferase mRNA (HGPRT).
Skeletal muscle cells were incubated under normoxic or hypoxic
conditions for 1 h, afterwards cells were collected and transcripts
were quantified by real-time PCR. Data were normalized using
glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) as housekeeping
gene. Data are shown as mean ± S.E. of three independent biological
experiments by triplicate. *=statistical differences between
treatments (p < 0.05).
Figura 2. Efecto de
la hipoxia en la expresión de las enzimas involucradas en el
metabolismo de purinas. A) Actividad de inosina monofosfato
dehidrogenasa (IMPDH), B) Actividad de purina nucleótido fosforilasa
(PNP), C) Actividad de Xantina oxidasa y D) hipoxantina guanina
fosforribosil transferasa (HPGRT) RNAm. Las células de músculo
esquelético fueron incubados bajo condiciones de normoxia o hipoxia
por 1 h, posteriormente las células fueron colectadas y los
trancritos cuantificados por PCR en tiempo real. Los datos fueron
normalizados usando gliceraldehído 3-fosfato dehidrogenasa (GAPDH)
como gen de referencia. Los datos representan medias ± D.S. de tres
experimentos biológicos independientes realizados por triplicado
cada uno. Los asteriscos denotan diferencias estadísticas entre
tratamientos (p < 0.05).
Purine metabolite concentration
The concentration of purine bases (HX, X), nucleosides (I, A), nucleotides (AMP, GDP, ADP, GTP, ATP), NAD+ and UA were measured by HPLC after normoxia and hypoxia treatments in human skeletal muscle cells (Table 2). UA was not detected in neither normoxic nor hypoxic cells. There was a reduction in the content of most of the metabolites, purine bases, nucleosides and nucleotides, in cells under hypoxic conditions as compared to normoxic cells; however, differences were only statistically significant for HX and IMP concentrations (p = 0.0031).
Table 2 Purine metabolite concentrations in human skeletal muscle cells exposed to hypoxia.
Tabla 2. Concentración de metabolitos de
purina en células de músculo esquelético de humano expuestos a
hipoxia.
| Metabolite | Normoxia | Hypoxia |
|---|---|---|
| HX | 1.54 (0.81, 2.17) | 0.76 (0.62, 0.99)* |
| Xanthine | 2.04 (0.99, 3.35) | 1.52 (0.98, 2.06) |
| UA | ND | ND |
| IMP | 1.04 (0.55, 2.12) | 0.42 (0.29, 0.62)* |
| Inosine | 4.71 (2.5, 7.22) | 3.33 (1.86, 4.38) |
| NAD+ | 0.25 (0.08, 0.50) | 0.21 (0.09, 0.78) |
| Adenosine | 0.18 (0.02, 8.34) | 1.11 (1.00, 1.15) |
| AMP | 0.18 (0.02, 8.34) | 1.11 (1.10, 1.15) |
| GDP | 0.07 (0.03, 0.11) | 0.05 (0.02, 0.08) |
| ADP | 0.37 (0.06, 7.17) | 0.09 (0.03, 2.68) |
| GTP | 4.56 (2.24, 6.88) | 0.08 (0.01, 2.20) |
| ATP | 0.31 (0.17, 0.87) | 0.16 (0.09, 0.60) |
Hypoxanthine (HX), xanthine, uric acid (UA), inosine monophosphate (IMP), inosine, nicotinamide adenine dinucleotide (NAD+), adenosine, adenosine monophosphate (AMP), guanosine diphosphate (GDP), adenosine diphosphate (ADP), guanosine triphosphate (GTP) and adenosine triphosphate (ATP). Data are expressed in µM mg-1 protein and represent five independent experiments for each treatment. Data are presented as median (25th-75th percentiles). ND = not detected. *=Significant differences between treatments, p < 0.05.
Hypoxia downregulates gene expression of enzymes that participate in purine metabolism
Transcripts encoding for IMPDH, PNP, XO and HPGRT were obtained from human skeletal muscle cells cDNA. The BLAST analysis of the sequences from the obtained fragments (~300 bp), confirmed the identity of the coding sequences obtained which correspond to the enzymes of purine metabolism. Quantitative real-time PCR was used to analyze the levels of expression of mRNAs encoding IMPDH, PNP, XO and HGPRT enzymes involved in purine metabolism (Fig. 2). IMPDH and XO were the most abundant transcripts, showing the highest significant reduction (p <0.01) after hypoxia in contrast to normoxic cells (~99% each). HGPRT mRNA showed a 58% reduction (p < 0.01) under hypoxic conditions, while PNP presented the lowest expression from all the transcripts analyzed, and it did not show significant differences in contrast to hypoxic cells.
Discussion
Mammals have developed effective strategies to cope with oxygen limiting situations. To achieve tolerance to acute or chronic hypoxia, the organism reduces metabolism, to prevent cellular injury and maintain functional integrity (Ramirez et al., 2007). Failure of these strategies results in different physiological and pathophysiological conditions (e.g. ischemia, inflammation and tumors) (Lahat et al., 2011).
The primary response to hypoxia within the cell ultimately leads to alternative routes of ATP generation (Wheaton and Chandel, 2011). IMPDH is a key enzyme in the purine nucleotide pathway, since it controls the gateway to guanine nucleotides (Hedstrom, 2009). IMPDH activity has been widely studied in several tissues, including plasma, erythrocytes, lymphocytes, spleen and thymus (Jonsson and Carlsten, 2001; Fukuda et al., 2011; Vethe et al., 2014; Citterio-Quentin et al., 2019) from different mammals and aquatic organisms (Velasco-Martinez et al., 2016). Differential activity patterns between tissues have been reported; low activity has been observed in erythrocytes and lymphocytes (89 pmol mg Hb-1 h-1, 4514 pmol mg-1 protein h-1, respectively) compared to human skeletal muscle cells in this study (46.59 µM mg-1 protein h-1). Human cells contain two IMPDH genes, which share 84% homology, although protein sequence differences does not affect their kinetic properties (Hedstrom, 2009). In the present study, a partial sequence of IMPDH from human skeletal muscle cells, amplified and sequenced contained a conserved functional region, shared by the two IMPDH genes. Normoxic cells showed high expression of IMPDH mRNA and IMPDH activity. After hypoxia treatment, IMPDH mRNA and IMPDH activity, as well as the endogenous levels of IMP, were downregulated, suggesting that oxygen limitation transcriptionally affects the de novo production of purines, as has been shown in normal lymphocyte cells (Jain et al., 2004) and hypoxia-treated human leukemic cells (Han et al., 2006).
PNP and XO are involved in purine degradation, PNP catalyzes the inosine breakdown to hypoxanthine (Dudzinska et al., 2006), the substrate for xanthine oxidase (Moriwaki et al., 1999). PNP activity has been reported in erythrocytes, liver and plasma of mammals (Schomaker et al., 2013; López-Cruz et al., 2014; Giuliani et al., 2016). Previous reports on PNP activity in human muscle cells show a higher activity in cell cultures than in human muscle homogenates (Jacobs et al., 1988). In this study, muscle cell PNP activity was 12.37 ± 2.71 U mg-1 under normoxic conditions, while after hypoxia treatment lower PNP activity was observed (6.79 ± 1.20 U mg-1), although this decrease was not significant. The same pattern was observed when the PNP mRNA was analyzed. In erythrocytes from rats, changes in PNP activity were evident after seven days of hypoxia (Hosek et al., 1986), while liver from the same species required an hour to stimulate its activity (Rao et al., 1989). In human adipocytes, PNP mRNA expression increased after 24h hypoxia (Nagao et al., 2018). The differential activity and expression pattern of PNP among tissues, might reflect different metabolic demands of each tissue and responses to stress conditions. XO activity in muscle tissue has been reported as less than 0.5 mU g-1 tissue (Dorion et al., 1993); in this study, higher levels of XO activity were obtained (17.55 ± 3.40 mU mg-1). The hypoxia treatment resulted in a mild increase of XO activity (21.44 ± 4.13 mU mg-1); however, this increase was not statistically significant. Previous reports indicate that moderate hypoxia significantly enhances XO activity in endothelial arterial cells (Kelley et al., 2006) and skeletal muscle and human plasma (Terada et al., 1992). The lack of increased activity in this study might be related to the significant reduction of HX levels observed after hypoxia (1.54 µM mg-1 under normoxia and 0.76 µM mg-1 under hypoxic conditions), limiting the catalytic capability of the enzyme. This level is consistent with the absence of UA and low levels of X. Interestingly, XO mRNA expression is modulated by hypoxia in a dose- and time-dependent manner, either by enhancing their expression (moderate hypoxia) or not affecting the gene expression (Poss et al., 1996; Kelley et al., 2006). In this study, XO mRNA in muscle cells was down-regulated in hypoxic conditions; these differences might relate to the time of exposure to hypoxia and to the level of regulation that XO presents, which is at transcriptional and posttranslational level (Kayyali et al., 2001; Poss et al., 1996).
The alternate pathway for purine nucleotide formation is the purine salvage pathway, in which instead of generating new purines, preexisting purine bases are taken up for the generation of nucleotides. HGPRT activity in human muscle cells has been previously reported to be 4.8 ± 0.015 nmoles mg-1 h-1 (Jacobs et al., 1988), which is in agreement with the levels obtained in the muscle cell culture under basal conditions (9.90 ± 3.6 nmoles mg-1 h-1) in this study. An increase of HGPRT activity has been observed after prolonged fasting (Soñanez-Organis et al., 2012) and as a compensatory response to hypoxia in mice heart tissue (Wu et al., 2015). Interestingly, cerebral hypoxia-ischemia experiments in rabbits did not show changes in HGPRT activity (Cherin et al., 2006). In this study, HGPRT activity decreased after hypoxia exposure; however, this reduction was not significant at the protein level, but it was significant at the transcriptional level.
The analysis of the purine metabolites showed a significant increase in HX content, which correlates with the low activity of XO observed. These results correlated with previous findings in adipocyte tissues exposed to hypoxia, which showed an increase in HX and acid uric secretion during prolonged hypoxia (Nagao et al., 2018). The significantly reduction of IMP concentration in this study, might be related to an active uptake of this compound by IMPDH, as has been shown in other mammalian tissues, including erythrocytes, placenta, liver and heart (Traut, 1994; Pérez-Milicua et al., 2015).
Conclusions
The observed reduction in enzyme activities for purine metabolism as well as their transcripts, along with the significant reduction of HX and IMP levels suggest that skeletal muscle cells respond differently to oxygen limitation than other tissues previously studied. This reduction might relate to a decrease of the metabolic machinery to avoid the excessive production of metabolites, which may lead to apoptotic signaling. This could suggest that skeletal muscle cells are able to withstand longer periods of hypoxia, before developing alternative mechanisms to cope with hypoxia effects via the salvage pathway, as has been observed in erythrocytes and plasma. However, further research is necessary to understand the mechanism underlying purine metabolism under hypoxia conditions.

