











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Vivimos una época inestable, caracterizada por ser altamente estresante y demandante. Percibimos distintos acontecimientos traumáticos de forma directa o indirecta (guerras, terrorismo, problemas económicos, robos/secuestros, accidentes de tráfico, violencia familiar o de pareja, feminicidios, e incluso catástrofes sobrenaturales y cambios climáticos, etc.), los cuales provocan secuelas y marcas emocionales que impactan en el desarrollo psicológico de las personas que los sufren. Entre los diversos trastornos que aparecen en la literatura, el más frecuentemente asociado al trauma es el trastorno de estrés postraumático (TEPT)1.
El TEPT es un problema de salud pública, cuya prevalencia alcanza entre un 5 y un 10%2 y se eleva considerablemente en poblaciones en riesgo (personal militar/policial, madres maltratadas o niños en situación de peligro/abuso, etc.). Recientemente, la Organización Mundial de la Salud desarrolló un estudio con la participación de más de 24 países, en el cual determinó que el 70.4% de los evaluados sufrían experiencias traumáticas a lo largo de su vida, con un riesgo y persistencia de TEPT de 77.7 años por persona de cada 100 sujetos3. Solo en Perú, la prevalencia del TEPT se sitúa entre el 14.6 y el 48.4%, variando en función de los grupos evaluados4,5.
El trastorno tiene lugar después que el individuo se enfrenta con un estrés severo o evento traumático que implica la amenaza de muerte o daño significativo a uno mismo o a un ser querido. Es el único trastorno neuropsiquiátrico que puede ser relacionado directamente con un evento desencadenante al cual los pacientes responden con miedo y angustia intensos4. Ha sido especialmente estudiado en veteranos de guerra y supervivientes a accidentes automovilísticos, además de reportes en violencia, abandono infantil, maltrato y abuso sexual5,6. Sin embargo, no todos los individuos que experimentan un acontecimiento traumático desarrollan TEPT.
Se asocia con altos niveles de discapacidad social, laboral y física7 y ocasiona considerables costes económicos y alta utilización médica. Los pacientes con TEPT suelen tener peor salud y mayores limitaciones en la vida diaria en comparación con otros cuadros mentales8,9, y existe una mayor presencia de intentos de suicidio y enfermedades físicas10.
Características clínicas del trastorno de estrés postraumático
El TEPT se caracteriza por la reexperimentación del trauma, comportamiento de evitación, alteraciones cognitivas y del estado del ánimo, así como por un incremento del arousal, que se manifiesta con irritación, hipervigilancia, sobresalto, alteraciones del sueño y pobre concentración7,9. Los pacientes muestran, además, sentimientos de ansiedad intensos y pueden revivir el evento traumático a través de los recuerdos intrusivos, flashbacks y pesadillas, por lo que evitan cualquier aspecto que les recuerda el trauma7. El trastorno posee una relación directa con el estatus metabólico, dado que suele presentar una alteración del eje hipotalámico-pituitario-adrenal (HPA) después del evento traumático11.
La comorbilidad en el TEPT es frecuente, así como la presencia de patología dual. Los trastornos mentales más prevalentes relacionados con el cuadro son la ansiedad, la depresión y el consumo de sustancias (alcohol, alucinógenos y psicotrópicos), los cuales alcanzan una comorbilidad de hasta un 50%, incluso 10 años después del primer diagnóstico de TEPT12.
Las variaciones individuales en la presentación clínica y/o preclínica del TEPT dependen de factores genéticos13, neurobiológicos, neuropsicológicos, cambios epigenéticos y de las influencias ambientales14. Sin embargo, a pesar de la perspectiva clínica fenomenológica y el incremento de la investigación básica de las últimas décadas, los procesos neurobiológicos subyacentes no se comprenden del todo15,16, siendo el objetivo de la revisión profundizar en el conocimiento de las características neurobiológicas más relevantes de los pacientes con TEPT.
Se realizaron búsquedas en diferentes artículos originales, incluidos estudios experimentales, clínicos y de revisión, en las bases de datos MEDLINE, PubMed y Science Direct, publicados después del año 2000. Las palabras clave de búsqueda fueron: «trastorno de estrés postraumático», «estrés postraumático», «neurobiología», «neurociencia» y «modelos animales», siendo seleccionados únicamente aquellos estudios relevantes en el análisis de la neurobiología del TEPT.
Desarrollo
Neurobiología del trastorno de estrés postraumático
Como otros trastornos neuropsiquiátricos, el TEPT representa un estado de enfermedad multifacético17, en el cual distintas variables juegan un papel en su desarrollo y neurobiología. Diversas áreas como la corteza cerebral, el hipocampo, la amígdala o el hipotálamo se muestran alteradas en el TEPT18 y se interconectan para formar circuitos neuronales que median distintas funciones como la adaptación al estrés, la toma de decisiones y el miedo condicionado. A continuación revisaremos brevemente algunas de estas estructuras.
CORTEZA PREFRONTAL
La corteza prefrontal dorsal, la corteza prefrontal medial (mPFC) y el área cingulada anterior (CAA) están implicadas en una variedad de funciones, entre las que se señalan los procesos cognitivos de alto nivel, como la función ejecutiva, la cognición social, la regulación emocional y la regulación del eje HPA19,20. Los estudios de cambios neurofisiológicos del TEPT se han centrado principalmente en el mPFC, ya que jugaría un papel significativo al mediar la inhibición de la amígdala18 y la modulación de la respuesta neurohormonal al estrés21. Respecto a lo último, se ha reportado que las neuronas de la mPFC contraen sus dendritas y muestran una menor ramificación19,22, además de mostrar potenciales a largo plazo alterados23. Los hallazgos se correlacionan con un menor rendimiento en pruebas de memoria de trabajo y función ejecutiva24, y esto jugaría un papel clave en el aprendizaje de extinción de miedo25. Un posible mecanismo que media estos cambios involucra la exposición excesiva a glucocorticoides y glutamato19, explicado en otro apartado.
Los estudios de neuroimagen han identificado también alteraciones funcionales y estructurales asociadas al trastorno. Por ejemplo, en un estudio realizado con veteranos de guerra diagnosticados con TEPT, se observó un espesor cortical frontal reducido en ambos hemisferios26. Además, la evaluación de memoria de trabajo y activación de la corteza prefrontal, utilizando espectroscopia funcional de infrarrojo cercano, ha encontrado que los pacientes tenían una menor actividad frontal durante los procesos de codificación y recuperación27 ante las tareas de memoria. En las misma línea, los estudios con resonancia magnética funcional muestran peor conectividad frontoparietal, que se relaciona con una peor ejecución de tareas con componente atencional y de funciones ejecutivas en estos pacientes16.
AMÍGDALA
La amígdala es una estructura vinculada con la emoción, con un papel crucial en la detección de amenazas y eventos emocionales28 relacionados con la adquisición y expresión del miedo condicionado. Esta estructura es capaz de perpetuar una respuesta de estrés mucho después de que un trauma ha terminado18,29, como se evidencia en un estudio realizado con ratas expuestas al olor de un depredador donde las células piramidales de la amígdala basolateral (BLA) mostraron una mayor longitud, número de dendritas y espinas30. Se ha observado también un incremento del flujo sanguíneo en la amígdala relacionada con una mayor ansiedad y síntomas de TEPT durante la adquisición del miedo, así como una activación incrementada ante la reexposición a estímulos con contenido traumático21,31.
HIPOCAMPO
El hipocampo es una importante estructura relacionada con la memoria, el aprendizaje y la emoción. Es el principal objetivo neural para los glucocorticoides por las altas concentraciones de sus receptores32. Los glucocorticoides hacen sensible al hipocampo a elevados niveles de estrés, aceleran el envejecimiento y la atrofia del área, lo que sumado a la reducción de factores neurotróficos se relaciona con menor neurogénesis, perdida de la ramificación neuronal, perdida de espinas dendríticas y atrofia21,30,32,33.
Estudios con humanos muestran menores volúmenes de hipocampo en personas diagnosticadas con TEPT21. Así, un estudio con imagen de resonancia magnética y con tomografía por emisión de positrones mostró que las mujeres que sufrieron abuso sexual en la infancia y desarrollaron TEPT tenían menor volumen del hipocampo (–16%) y menor activación del área asociada a la memoria declarativa (–19%,) en comparación a mujeres que sufrieron abuso pero no desarrollaron el trastorno34.
La disfunción de las subregiones del hipocampo mediaría la sobregeneralización del miedo y los recuerdos intrusivos7,35; es el caso del hipocampo anterior, relacionando la disminución de la conectividad funcional con la corteza prefrontal y CAA con una mayor presencia de sintomatología de TEPT36.
HIPOTÁLAMO E HIPÓFISIS
El hipotálamo inicia distintos ejes de regulación metabólica y homeostática de gran importancia y complejidad, además de mediar la respuesta al estrés por medio del eje HPA37. Frente a un estresor, el núcleo paraventricular del hipotálamo sintetiza la hormona liberadora de corticotropina (CRH). La CRH se libera hacia los vasos portales que acceden a la hipófisis anterior para unirse a su receptor e inducir la liberación de la hormona adrenocorticotropa (ACTH). El objetivo principal de la ACTH circulante es la corteza suprarrenal, donde estimula la síntesis de glucocorticoides al unirse a los receptores de mineralocorticoides en la corteza adrenal38.
El producto final de la actividad del eje HPA es el cortisol en humanos o la corticosterona en roedores39. El cortisol tiene una influencia directa en los circuitos cerebrales implicados en la homeostasis de la emoción, y a su vez controla la actividad del eje HPA. La respuesta al estrés ligada a la regulación del feedback negativo en el HPA estaría mediada por los receptores de glucocorticoides, mientras que los receptores de mineralocorticoides se encargarían de los estados basales del eje HPA (Fig. 1).
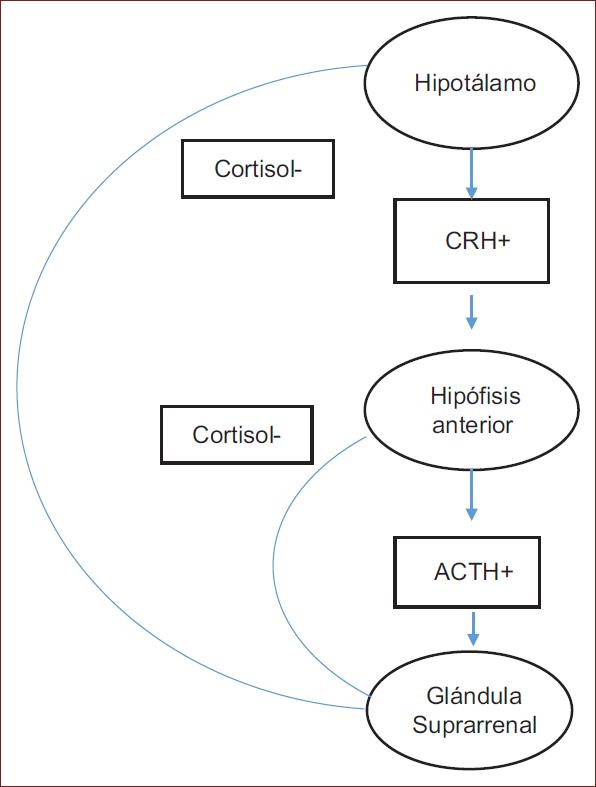
EL EJE HIPOTALÁMICO-HIPOFISARIO-ADRENAL EN EL TEPT
La actividad normal del eje HPA conduce a la liberación rítmica y episódica de glucocorticoides suprarrenales, proceso esencial para la homeostasis del cuerpo y la supervivencia durante el estrés, además de promover las respuestas de regulación de vías metabólicas, cardiovasculares, actividad inmunológica y neuroendocrina42. Los glucocorticoides cumplen la función de liberar sustratos de energía y así asegurar su disponibilidad en la oxidación mitocondrial para la respuesta de lucha o huida. Su sobreexposición altera el metabolismo afectando la acción de la insulina y generando su resistencia41.
Por otro lado, estudios iniciales mostraron discrepancias sobre los niveles de cortisol en el TEPT42. La evidencia señala niveles reducidos de cortisol durante el día y niveles elevados al anochecer43-45; es el caso del estudio desarrollado por Griffin, Resick y Yehuda46, donde los niveles de cortisol basales a las 9 a.m. mostraron una reducción significativa en mujeres con TEPT que sufrieron violencia doméstica. Datos similares se han encontrado en sujetos que sufrieron abuso sexual en la infancia43, así como en veteranos de guerra diagnosticados47.
Lo referido sugiere que la alteración del eje HPA es una característica predominante de la fisiopatología del trastorno. Existen una amplia gama de condiciones que pueden alterar el eje HPA48 o sus receptores49, como polimorfismos genéticos, cambios en la expresión de genes por factores epigenéticos y condiciones ambientales, entre otras50.
Test de supresión de dexametasona
La dexametasona es un glucocorticoide sintético con acciones agonistas a los glucocorticoides, tiene una afinidad entre 20-30 veces mayor al cortisol y la corticosterona. Su mecanismo de acción incluye la supresión de la CRH y la ACTH51 por medio de la unión a los receptores de glucocorticoides y provoca la activación del feedback negativo sobre la liberación de corticosterona. Además, muestra un limitado acceso al cerebro debido a la barrera hematoencefálica52. Su distribución y acción selectiva se da especialmente en la hipófisis, a través del sistema portal. En el TEPT, una supresión mayor del cortisol en respuesta a la dexametasona refleja un aumento de la respuesta de inhibición del eje HPA y sería reflejo del incremento en la sensibilidad de sus receptores48,53.
El test de supresión de dexametasona (TSD) ha sido ampliamente utilizado para evaluar el eje HPA en diversos trastornos psiquiátricos52. Es reconocido como medida para la detección de alteraciones funcionales en el sistema HPA por su fácil uso y su capacidad de detección de cambios fisiopatológicos. Ha sido usado en cuadros como la esquizofrenia, depresión y ansiedad, además de actuar como marcador biológico para el pronóstico de la respuesta del TEPT frente a la psicoterapia. Es el caso de Nijdman, et al., quienes reportaron un incremento de los niveles de supresión de cortisol, que se relacionan con mayor sintomatología y peor pronóstico, siendo las medidas de sensibilidad del eje HPA predictoras de la respuesta clínica positiva54.
También se han observado los efectos del TSD frente al miedo condicionado. Sus resultados sugieren que la supresión transitoria de la función HPA puede reducir el miedo exagerado en pacientes con TEPT55. Dosis bajas de dexametasona producen una activación selectiva de los receptores de glucocorticoides en la hipófisis mientras que no muestran una acción similar en el tejido cerebral52. Este uso concuerda con la idea de emplear glucocorticoides o inhibidores del receptor de glucocorticoides para potenciar la actividad del eje HPA como tratamiento en el TEPT53.
Alteración del sistema nervioso autónomo en el TEPT
Distintos estudios han abordado las alteraciones del sistema nervioso simpático (SNS) enfocando su atención en la hiperactivación y excesiva respuesta adrenérgica en el TEPT28. Las proyecciones noradrenérgicas que parten desde el locus cerúleo hacia la corteza prefrontal, la amígdala, el hipocampo y el hipotálamo, muestran una actividad incrementada ante un estresor56. Esta actividad moviliza recursos energéticos para la respuesta de lucha o huida. La noradrenalina, por ejemplo, aumenta la frecuencia cardíaca y el flujo de sangre a los músculos esqueléticos, y provoca la liberación de la glucosa. A su vez, desde el hipotálamo lateral, el sistema orexinérgico promueve la activación del locus cerúleos, propiciando cambios en la regulación de estado de sueño/vigilia, la emoción y la cognición en respuesta al miedo57.
En el TEPT, el SNS se caracteriza por una excesiva actividad noradrenérgica y esto se relaciona con mayor incidencia de enfermedad cardiovascular, mayor presencia de síntomas de TEPT y síndrome metabólico58. Además de asociarse a un respuesta de hipervigilancia, atención selectiva e hiperarousal59. La noradrenalina tiene un papel sustancial en la memoria a través de los glucocorticoides. Cuando la actividad noradrenérgica aumentada se asocia a la reducción de los glucocorticoides en la BLA, la respuesta al miedo condicionado se incrementa60. El eje HPA influencia también la actividad de la noradrenalina, mediante el incremento de CRH56. Esta respuesta implica el mantenimiento de los síntomas del TEPT como hiperactivación del SNS, refuerzo del miedo condicionado, pesadillas, flashbacks, etc.
Sistemas de neurotransmisión implicados en el TEPT y el estrés
Se ha observado que distintos sistemas de neurotransmisión se encuentran alterados en el TEPT. La presencia de un estresor impredecible durante el desarrollo aumenta los comportamientos de ansiedad en la edad adulta y se producen cambios en genes relacionados con la neurotransmisión de glutamato, ácido gamma aminobutírico (GABA), noradrenalina y serotonina, así como en sus respectivos receptores61. El condicionamiento y extinción al miedo se encuentran alterados en el TEPT y son mediados por el receptor de N-metil-d-aspartato (NMDA), en la amígdala y la mPFC62. Las alteraciones de la plasticidad sináptica, retracción dendrítica y pérdida de espinas en la mPFC están asociadas con un exceso de activación de los receptores de NMDA por el aumento de los niveles de glutamato63; efectos similares se encontraron en el hipocampo de roedores sometidos a estrés agudo y severo64.
El glutamato en exceso en el cerebro provoca una amplia respuesta proinflamatoria y excitotoxicidad. En sujetos con TEPT se encontraron mayores niveles de glutamato en suero, menores ratios de glutamina/glutamato, asociados inversamente con la sintomatología del trastorno, y menores niveles de cortisol en saliva65. Sin embargo, el glutamato no se libera de forma aislada, algunos sistemas como el de la orexina lo utilizan de cotransmisor (actividad de orexina/glutamato, además de orexina/neurotensina, orexina/dinorfina, etc.), para generar una respuesta ante eventos de pánico, miedo o la regulación de la percepción del dolor y la analgesia66.
Por otro lado, un desequilibrio entre el GABA y el glutamato provocarían una serie de cambios en la expresión de síntomas del TEPT. El incremento del glutamato puede promover la apoptosis en neuronas del hipocampo y la mPFC propiciando la patogénesis del TEPT64. Así mismo, los niveles disminuidos de GABA en suero67 podrían relacionarse con el estrés, la hiperexcitabilidad sináptica y la aparición de crisis convulsivas psicogénicas68,69.
Modelos animales en el estudio del TEPT
Para un estudio más detallado de las alteraciones neurobiológicas del TEPT se realizan distintos modelamientos del trastorno en animales, utilizando para tal fin la exposición a diversos estresores (inmovilización, nado forzado, footshock, inestabilidad del alojamiento, separación materna, etc.). No obstante, el abordaje de los mecanismos implicados en el desarrollo del TEPT requiere el estudio de modelos animales específicos que incorporen validez aparente y de constructo. Dichos modelos reflejan la sintomatología del TEPT de acuerdo con los hallazgos y características teóricas ya encontradas, así como las alteraciones neurobiológicas y neuroendocrinas, permitiendo una mayor comprensión de su fisiopatología28.
Dentro de los modelos animales específicos de TEPT destacan la exposición a depredador (ED), el estrés prolongado único (SPS, single prolonged stress) y el choque eléctrico con estresores adicionales28,70, los cuales mimetizan mejor las distintas características del TEPT. A continuación, describiremos cada uno de ellos:
–En el modelo de ED, los animales son expuestos a un depredador natural o a su olor en un medio en el que no pueda escapar28, reflejando el daño potencial al que se ven expuestos las personas que desarrollan TEPT70. Los animales sometidos a los eventos simulan diversos criterios diagnósticos de TEPT, entre los que se observan una mayor ansiedad, una respuesta de sobresalto exagerada y déficits de memoria50, además de freezing y conducta de evitación17. Los síntomas persisten en el tiempo algunas semanas o incluso meses13. El modelo de ED refleja también las alteraciones en hipocampo, amígdala y mPFC,30 así como la desregulación del eje HPA, del sistema nervioso simpático17 y del sistema inmunitario, mostrando un incremento de la respuesta inflamatoria, con mayores niveles de TNF-α, IL-6 y CD45 (un marcador de actividad microglial) en hipocampo e hipotálamo71.
–El SPS, contrariamente a su nombre, implica el uso de múltiples estresores, como la inmovilización durante dos horas, nado forzado durante 20 minutos y la exposición a éter hasta la pérdida de consciencia. Este modelo reproduce la alteración del eje HPA, con una disminución de los niveles de corticosterona, así como una sobreexpresión de los receptores de glucocorticoides70. Se ha observado que representa también otros síntomas asociados al TEPT como la hiperexcitación, freezing y ansiedad19.
–El modelo de footshock con estresores adicionales utiliza un único episodio de descarga eléctrica asociado a un estímulo condicionado o recordatorios contextuales28. Dicho modelo muestra un incremento de la respuesta de sobresalto, hipersensibilidad noradrenérgica, disminución de la conducta social, inmovilidad, neofobia, etc. El uso de recordatorios contextuales incrementa la agresividad y la respuesta de sobresalto, además de disminuir la conducta exploratoria71.
Finalmente, los modelos animales del TEPT permiten realizar un estudio detallado de las alteraciones neurobiológicas del trastorno, neurológicas, del sistema inmunitario, endocrinas y metabólicas, donde estas últimas estarían ligadas a la alteración de factores neurotróficos, en relación a la actividad regulatoria del hipotálamo72.
Conclusiones
En esta breve revisión se han abordado los aspectos más relevantes ligados a la neurobiología del TEPT, un trastorno muy particular en la clínica debido a su distintiva característica de ser rastreable hasta un evento traumático en la vida del paciente. Este hecho es una herramienta única, debido a que se pueden estudiar los cambios en el cerebro con investigación en neurociencia básica y el modelamiento del trastorno en animales. Los mismos que, sumados a los datos aportados por pacientes, confirman una marcada alteración de la actividad prefrontal y cingulada (reducción de la corteza cerebral, disminución de dendritas e hipoactivación), de la amígdala (hiperactivación) y el hipocampo (especialmente atrofia).
Los cambios producto del TEPT se relacionan directamente con la actividad neurocognitiva de los pacientes, quienes muestran principalmente una alteración de las funciones ejecutivas (atención, memoria de trabajo, planificación, etc.) y de la regulación del estado del ánimo. Sin embargo, la alteración provocada por el trastorno no se limita a los procesos cognitivos y a sus estructuras subyacentes. Existen además zonas como el hipotálamo, relevante para el control de la homeostasis metabólica y endocrina del sujeto, que muestra cambios por medio de la actividad disfuncional del eje HPA, de los glucocorticoides (cortisol en sangre o saliva) y sus respectivos receptores (resistencia o disminución de receptores disponibles en áreas reguladoras como el hipocampo o el hipotálamo). Los cambios metabólicos alteran a su vez la capacidad de conservación y protección del cerebro, modificando el funcionamiento de distintos factores neurotróficos, muchos de ellos ligados al correcto funcionamiento del hipotálamo.
Se debe de tener en cuenta que la mayoría de los trastornos neuropsiquiátricos no comportan la alteración específica de una red neural, área o sistema, sino que en su conjunto presentan una alteración de distintos sistemas simultáneamente, que se solapan y a su vez se hacen evidentes con endofenotipos neurocognitivos relativamente específicos (funciones ejecutivas, toma de decisiones, teoría de la mente, coherencia central, cognición social, etc.). Sin embargo, su aproximación neurocognitiva aportará luces sobre los sistemas neurobiológicos alterados, y la investigación básica y la utilización con modelos animales proporcionaría los conocimientos de la alteración celular y molecular relacionada, aspectos significativos en la evaluación, tratamiento y seguimiento de estos pacientes.
Los actuales acontecimientos que sufre nuestra sociedad marcan la pauta de la salud mental de la población. Es por ello que debemos tener en cuenta el papel multifacético de los trastornos mentales (biopsicosocial) y cómo alteran las distintas actividades de la vida diaria de quienes sufren directa o indirectamente cada uno de estos problemas. A menudo, la población general presenta sintomatología subclínica que pasa desapercibida en atención primaria y que no recibe tratamiento oportuno durante años. Dicha situación genera un desborde de la capacidad de control mental y regulación emocional, soportando una problemática psicosocial relacionada (revictimización, violencia, desajuste socioemocional, suicidio, etc.). Por ello es importante la intervención multidisciplinaria, así como el desarrollo de políticas de salud pública en el ámbito socioemocional que lleguen a las poblaciones más vulnerables; un aspecto que desde la psiquiatría y la psicología sociocomunitaria, y de intervención en emergencias, es necesario promover.

