











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Monoprotection of symmetrical or unsymmetrical diamines is an ongoing challenge to organic chemists. Many of these monoprotected chiral 1,2-diamines have been used in organocatalysts and ligand-metal catalysts for many asymmetric transformations, such as asymmetric transfer hydrogenation. Aldol reaction, Michael, reaction silylcyanation, and Strecker reaction [1-7]. From these diamines, non-racemic trans-cyclohexane-1,2-diamine (1) is readily available in both enantiomeric forms, by simple resolution of the commercial crude racemate with tartaric acid [8]. Several methods for monoprotection of diamine (1) have been reported in the literature (Fig. 1) [9]. Some of these methods report excellent yields of the monoprotected diamine; yet, others in our hands led to mono- and di-protected product formation needing extensive chromatographic separation.

Results and discussion
A recent report highlighted the use of acetyl chloride to monoprotect cyclohexane-1,2-diamine at 0 °C, whereas in our hands the main product obtained was the diprotected cyclohexane-1,2-diamine (9b). However, the reaction of the diamine with various arylsulfonyl chlorides give mainly to the monoprotected sulfonylamide. Recently we employed a wide variety of these enantioenriched monosulfonamides in the asymmetric transfer hydrogenation (ATH) of aromatic ketones (1a) [10].
All of these ligands were readily synthesized by reacting (1R,2R)-cyclohexane-1,2-diamine tartrate salt with arylsulfonyl chloride in DCM/NEt3 at 0 °C. The monosulfonamides were obtained in ~40% yields using 5 fold excess of the diamine, with little diprotected amine formation, which can be purified by acid workup [11]. Literature also reveal that sulfonamides with electron withdrawing groups, such as nitro can be deprotected using PhSH/K2CO3 in DMF. Since these monosulfonamides can be prepared easily, we explored the deprotection of p-NO2 (1e) and 2,4-dinitro (1f) arylsulfonamides using the above mentioned methods. Unfortunately, all attempts led to a mixture of products (Fig. 2).

Looking for an efficient and simple method for diamine monoprotection, we found the procedure developed by Ha and co-workers, involving the monoprotonation of the diamine 1 using 1 equivalent of HCl, followed by treatment with Boc all in one pot, led to 80 % of mono-Boc protection of diamine (1a) [12]. Similary mono hydrochlorination was also reported by Nguyen in the synthesis of mono-diamine Schiff bases [13]. Li and coworkers reported preparation of mono-Boc protected mixed primary-secondary amines starting with a bromoalkylamine hydrobromide and Boc2O/Et3N in MeOH with good yields [14]. There are many literature citations to make mono-Boc protected cyclohexane-1,2-diamine using the mono-hydrochlorination method [13], the only drawback of this reaction is the use of compressed anhydrous HCl gas. An alternative HCl source is the in situ generation of HCl from chlorotrimethylsilane ((Me)3SiCl) or thionyl chloride (SOCl2) with anhydrous methanol. Chlorotrimethylsilane, a liquid with a boiling point of 57 °C can be purchased in >99% purity and redistilled before use [15], and can be weighed accurately to generate the required amount of HCl gas with anhydrous methanol.
The methodology proposed (Fig. 3) consisted in treating (1R,2R)-cyclohexane-1,2-diamine tartrate salt with 4N NaOH, to give the diamine (1) as a free base. Then methanol anhydrous at 0 °C is added, followed by dropwise addition of 1 eq of Me3SiCl. The mixture is allowed to come to RT and 1 mL of water is added, followed by Boc2O in MeOH. The mixture is stirred at RT for 1 hr, diluted with water, washed with ethylic ether, pH adjusted to >12 with NaOH and then extracted into dichloromethane to give a pure monoprotected cyclohexane-1,2-diamine (1a) in 66% yield (Table 1). We were able to scale up the methodology to ~4.0 g.
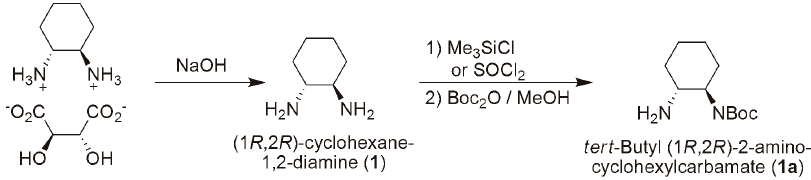
The monoprotection was also carried out with SOCl2 as the HCl source using the same work-up procedure as above, but the final product was obtained in lower yield (41%).
This methodology was also used with 1R,2R-1,2-diphenylethyl-1,2-diamine (2) and seven aliphatic diamines (3-9). Following the same procedure as obtaining 1a, we were able to obtain the mono-Boc protected diamines 2a-9a (Fig. 4) in moderate yields (Table 1). For the racemic tert-butyl (2-aminopropyl) carbamate (3a), the monoprotection goes to the amine at C-1, suggesting that 1 eq of Me3SiCl as HCl source, prefers to protonate amine C-2, that is more basic than the amine at C-1, leaving the monoprotection at C-1. This was established by analysis of NMR HMBC experiment were the protons of the methylene (δH 3.15 and 2.90 ppm) correlate with the carbamate carbonyl (δC 156.3 ppm), confirming the monoprotection on the C-1 amine. Purity was determined by GC-MS and all monoprotected diamines 1a-9a were obtained from 93 up to >99% pure (Table 1). Spectroscopic data were in good agreement with those reported in the literature [9d, 13c, 16-20].

In conclusion, we have reported an efficient and a simple “one-pot” reaction for obtaining mono-Boc protected diamines (1a-9a) using Me3SiCl to generate the mono HCl salt of the diamines. This method can be extended to other diamines.
General Experimental Procedures
Melting points were obtained on an Electrothermal 88629 apparatus and are uncorrected. Infrared spectra (IR) were recorded on a Perkin Elmer FT-IR 1600 spectrophotometer. NMR spectra were recorded on a Bruker Avance III spectrometer 400 MHz, in CDCl3 with TMS as internal standard. Mass spectra were obtained on an Agilent Technologies 5975C MS Spectrometer at 70 eV by direct insertion.
General procedure of mono-Boc protected of diamines 1a-9a
Each diamine 1-9 (1 eq ≈1gr) was added anhydrous methanol at 0 ºC under stirring, followed by the dropwise addition of freshly distilled Me3SiCl (1 eq). A white precipitate appeared at the bottom of the flask, the mixture was allowed to come to RT and water (1 mL) followed by Boc2O (1 eq) in MeOH (3 mL) was added. The mixture was stirred at RT for 1 hr, diluted with water (50 mL) and the aqueous layer washed with ether (2 × 75 mL). The aqueous layer was adjusted to pH >12 with 2N NaOH and extracted into dichloromethane (3 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4 and solvent removal gave the corresponding monoprotected diamines 1a-9a.
tert-Butyl (1R,2R)-2-aminocyclohexylcarbamate (1a) as a white solid 1.97 g, 66% yield. [α]20 D= -44° (c 3.0 mg/mL, MeOH). M.p. 105-107 ºC; IR: 3351, 2909, 2888, 1882, 1518, 1239, 1166, 1015, 983 cm-1. 1H NMR (400 MHz, CDCl3): δ 4.49 (brs, NH), 3.13 (brd, J = 6.2 Hz, 1 H), 2.33 (ddd, J = 10.4, 3.8, 3.8 Hz, 1H), 1.98 (m, 2H), 1.70 (m, 2H), 1.45 (s, 9H), 1.28 (m, 2H), 1.12 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 156.1, 79.4, 57.6, 55.7, 35.2, 32.9, 28.4, 25.2, 25.1. EIMS m/z: [M]+ 214 N.D., 157 (1), 141 (13), 114 (8), 97 (100), 70 (15), 56 (47). Spectroscopic data were in good agreement with those reported in the literature (9d, 13c, 16).
The monoprotection for 1, was also carried out with SOCl2 (6.7mmol) as the HCl source (caution! reacts vigorously with methanol). The addition was carried out at -200 C. Using the same work-up procedure as above, the final product tert-Butyl (1R,2R)-2-aminocyclohexylcarbomate (1a) was obtained in 1.24g, 41%.
tert-Butyl ((1R,2R)-2-amino-1,2-diphenylethyl)carbamate (2a) White solid (665 mg, 45%); 1H NMR (400 MHz, CDCl3): δ 7.32-7.24 (m, 10H), 5.78 (brs, NH), 4.85 (brs, NH2), 4.33 (d, J = 4.2 Hz, 1H), 4.09 (s, 1H), 1.32 (s, 9H). 13C RMN (100 MHz, CDCl3): δ 155.7, 143.4, 142.3, 128.5, 128.3, 128.2, 127.4, 127.2, 127.0, 126.9, 126.8, 126.5, 79.3, 61.9, 60.0, 28.3. EIMS m/z: [M]+ 312 N.D., 238 (2), 222 (3), 196 (2), 150 (7), 106 (100). Spectroscopic data were in good agreement with those reported in the literature [17].
tert-Butyl (2-aminopropyl)carbamate (3a) White solid (1.7 g, 72%), M.p. 68-70 ºC; 1H NMR (400 MHz, CDCl3): δ 7.69 (brs, NH), 5.07 (brs, NH2), 3.14 (brs, 1H), 3.02 (m, 1H), 2.90 (m, 1H), 1.45 (s, 9H), 1.08 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 156.4, 79.2, 47.8, 47.1, 28.4, 20.1. EIMS m/z: [M]+ 174 N.D., 101 (24), 75 (11), 57 (100), 44 (100). Spectroscopic data were in good agreement with those reported in the literature [18].
tert-Butyl (2-aminoethyl)carbamate (4a) White solid (587 mg, 22%), M.p. 64-66 ºC; 1H NMR (400 MHz, CDCl3): δ 5.55 (brs, NH), 4.54 (brs, NH2), 3.24 (d, J = 5.2, 2H), 3.15 (brs, 1H), 2.86 (brs, 1H), 1.44 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 156.3, 79.3, 41.8, 41.1, 28.4. EIMS m/z: [M]+ 160 N.D., 118 (7), 103 (5), 87 (52), 75 (44), 57 (100), 43 (49). Spectroscopic data were in good agreement with those reported in the literature [18].
tert-Butyl (3-aminopropyl)carbamate (5a) White solid (565 mg, 24%), M.p. 64-66 ºC; 1H NMR (400 MHz, CDCl3): δ 5.33 (brs, NH), 4.95 (brs, NH2), 3.21 (d, J = 5.6 Hz, 1H), 3.11 (brs, 1H), 2.82 (t, J = 6.4 Hz, 2H), 1.70 (quint, J = 6.6, Hz, 2H), 1.43 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 156.3, 79.1, 38.4, 37.9, 31.3, 28.5. EIMS m/z: [M]+ 174 (1), 118 (51), 101 (64), 74 (20), 57 (100). Spectroscopic data were in good agreement with those reported in the literature [18].
tert-Butyl (4-aminobutyl)carbamate (6a) White solid (406 mg, 19%), M.p. 64-66 ºC; 1H NMR (400 MHz, CDCl3): δ 4.89 (brs, NH), 3.11 (brs, 4H, CH2, NH2), 2.75 (t, J = 6.4 Hz, 2H), 1.52 (m, 4H), 1.44 (s, 9H). [13]C NMR (100 MHz, CDCl3): δ 156.1, 79.1, 41.3, 40.3, 29.8, 28.5, 27.4. EIMS m/z: [M]+ 188 (1), 132 (52), 115 (47), 103 (30), 80 (9), 70 (84), 57 (100). Spectroscopic data were in good agreement with those reported in the literature [18, 19].
tert-Butyl (6-aminohexyl)carbamate (7a) White solid (781 mg, 42%), M.p. 67-69 ºC; 1H NMR (400 MHz, CDCl3): δ 4.60 (brs, NH), 3.03 (q, J = 6.0 Hz, 4H), 2.62 (t, J = 7.2, 6.4 Hz, 2H), 2.47 (brs, NH2), 1.37 (brs, 4H), 1.36 (brs, 13H), 1.26 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 156.0, 79.0, 41.7, 40.5, 32.9, 30.0, 28.5, 26.6, 26.5. EIMS m/z: [M]+ 216 (2), 159 (19), 143 (59), 131 (37), 98 (46), 86 (71), 57 (100). Spectroscopic data were in good agreement with those reported in the literature [18].
tert-Butyl (7-aminoheptyl)carbamate (8a) White solid (814 mg, 46%), M.p. 39-41 ºC; 1H NMR (400 MHz, CDCl3): δ 4.50 (brs, NH), 3.02 (q, J = 6.4 Hz, 2H), 2.61 (t, J = 7.0 Hz, 2H), 1.56 (brs, NH2), 1.37 (brs, 13H), 1.24 (brs, 6H). 13 C NMR (100 MHz, CDCl3): δ 155.0, 78.0, 41.1, 39.6, 32.5, 29.0, 28.1, 27.4, 25.8, 25.7. EIMS m/z: [M]+ 230 (2), 173 (23), 157 (51), 145 (31), 112 (28), 100 (54), 57 (100). Spectroscopic data were in good agreement with those reported in the literature [19, 20].
tert-Butyl (8-aminooctyl)carbamate (9a) White solid (711 mg, 42%). 1H NMR (400 MHz, CDCl3): δ 4.51 (brs, NH), 3.03 (q, J = 6.3, 6.0 Hz, 2H), 2.62 (t, J = 7.0 Hz, 2H), 2.22 (brs, NH2), 1.37 (brs, 13H), 1.23 (brs, 8H). 13C NMR (100 MHz, CDCl3): δ 155.0, 78.0, 40.9, 39.6, 32.2, 29.0, 28.3, 28.2, 27.4, 25.8, 25.7. EIMS m/z: [M]+ 249 (4), 187 (25), 171 (58), 143 (19), 114 (35), 57 (100). Spectroscopic data were in good agreement with those reported in the literature [19, 20].
General protocol for monitoring Mono-Boc Protection of diamines by gas chromatography-mass spectrometry (GC-MS)
The analytical GC/MS system used was an Agilent 7890A GC coupled to 5975C Mass detector Agilent Technologies, equipped with a HP-5MS capillary column (30 m x 0.25 mm x 0.25 μm) Agilent Technologies, Inc. An Agilent Technologies 7693 auto sampler was used to inject 1 μL of a solution sample. The ionization energy was 70 eV with a mass range of 30 to 800 m/z. The initial temperature of the column was set at 70 °C, held for 2 min, and then a ramp of 40 °C/min to 250 °C. The temperature of the injector was set at 250 °C, and the detector at 230 °C. The flow rate of the carrier gas (Helium) was 1.0 mL/min injected with a gas dilution of 1:50. Identification of the individual components was based on comparison with the mass spectra library (NIST98).

