











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
The design and preparation of salts or co-crystals of API’s (active pharmaceutical ingredients) is a current trend in Crystal Engineering and pharmaceutical sciences. It is well known that physical chemical and biological properties of API’s can be altered incorporating co-formers without having any covalent modification forming co-crystals [1]. Among these pharmaceutical co-crystals, there is a great interest in Metformin due to its global importance as an oral antidiabetic drug. Metformin is a biguanidine compound used for the treatment of type 2 diabetes mellitus (T2DM), in particular in overweigh and obese people. Metformin hydrochloride (MET·HCl) 1 improves glucose tolerance in patients with T2DM, lowering plasma glucose levels [2] and glycated hemoglobin HbA1c. [3-5]
Despite these benefits 1 is associated with gastric disorders in patients with prolonged use due to the release of high concentration of chloride anion [6] and metformin freebase has low intestinal absorption (approximately 50% to 60%) [2]. Fact that has encouraged the scientific community to design and prepare new Metformin pharmaceutical based formulations [6].
In this context, we explored the preparation of [MET]2 [SUC] 2 by melt mixing of 1 in the presence of dehydrated Na2[SUC]2. In this case, we are interested in replacing the anion Cl- based on the concepts of the crystal engineering and using melting procedures [7], exploiting the well-known ability of the carboxylate and guanidinium groups to form charged-assisted hydrogen bonding in solid state (Scheme 1) [8].
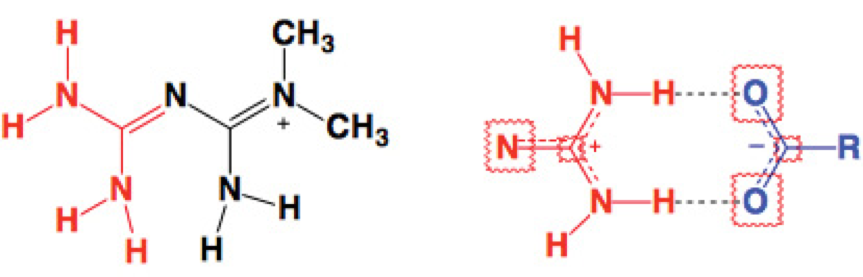
Scheme 1 H-MET+, guanidinium moiety (left, red), guanidinium(red)...carboxylate(blue) dimer motif assisted by charge (right).
According to Nanubolu et. al. [8] ten metforminium (H-MET+) salts with different anions have been deposited at the Cambridge Structural Database CSD [9]. Five of them including the monocation H-MET+ (two hydrochloride polymorphs, bromide, nitrate and one acetate salts) and five more having the metformin dication (H2-MET2+, oxalate, sulfate, perchlorate, squarate and calixiarenesulfonate salts). Nanubolu also discusses the formation of two polymorphs of the salt [MET]2[EMB] (EMB2- = embonate) [8]. In addition, the formation of the salt [MET][SAL] (SAL- = salicylate) [10], and the drug-drug salt [MET][GLI] containing two antidiabetic agents in the same solid phase (GLI, glicazide is a drug of the sulfonylurea-type) have been recently reported [11].
Some patents related with formation of organic salts, including at least one H-MET+ and one carboxylate group are: (a) US 006031004 A, salts of [MET]2[SUC] and [MET]2[FUM] (FUM2- = fumarate) only preparation by dissolution processes and thermal behavior [12a-b]. (b) WO 2009/038396 A2, which describes the formation of a family of salts of H-MET+ with a series of carboxylates [malate (MAL2-), malonate (MLN2-), glutarate (GLU2-) and adipate (ADI2-)] [12c-d]. (c) WO 2008/093984 A1, salt of [MET][ACE] (ACE- = acetate) [12e]. (d) US 2014/0249221 A1, describes the formation of salts containing: H-MET+, eicosapentaenoate/docosahexaenoate and aspartate or glutamate [12f]. (e) WO 2009/085198 A2, salt of H-MET+ with α-lipoate, combined with different anti-obesity agents [12g]. (h) WO 2009144527 A1, salt of [MET][GLY] (GLY- = glycinate) [12h] and (i) KR 2011123881 A, salt of [MET]2[SUC] [12i].
The salt of 2 has been extensively studied for its biological effects; however, its crystalline structure and solid state NMR spectroscopic characterization has not been reported. Thus, the results obtained from the synthesis by melting of the pharmaceutical salt 2 including its X-Ray crystal structure, spectroscopic studies and physical chemical properties are summarized below.
2. Experimental
2.1 Materials and Methods
All reagents for synthesis and analysis were of analytical grade and were used as received without further purification. MET·HCl 1 was purchased from Tokyo Chemical Industry Co. All other reagents were obtained from Sigma Aldrich Chemical Co. and the solvents were purchased from JT Baker. The disodium succinate salt was obtained by vigorous grinding (agate mortar and pestle) of succinic acid (H2SUC) in the presence of NaOH (1:2 molar ratio). The reaction is exothermic and water is produced during the process affording a white paste. This paste was further grinded for 10 min. affording a white powder that were desiccated in an oven at 110º C for 4 h.
Na2[SUC]. IR-ATR (cm-1). 3361 (w), 3250 (w), 1669 (m), 1544 (s), 1401 (s), 1237 (w), 1157 (w), 816 (m), 729 (s), 663 (s), 552 (s). ssCPMAS 13C NMR(ppm). Na+-OOC-*CH2-*CH2-COO-Na+ (δ = 34.74), Na+-OOC*-CH2-CH2-C*OO-Na+ (δ= 181.60).
2.2 Synthesis by Melt Mixing
MET·HCl, 1 (165.6 mg, 1.0 mmol) and dehydrated disodium succinate (81 mg, 0.5 mmol) were melted at 220 °C for 5 min. and the resulting white powder was ground for 10 min at room temperature. Suitable single crystals of 2 for X-Ray diffraction were obtained by slow evaporation of saturated methanolic of 1 at room temperature. Figures of the crystalline structures of 2 were generated by the programs X-Seed [13], and Olex 2 [14].
2.3 Spectroscopic characterization and thermal analyses
Infrared spectra were recorded on a Bruker Tensor 27 ATR spectrophotometer and measured in the range of 4000-400 cm−1. Melting points of 1 and 2 were determined using a Fisher Johns apparatus and are reported without correction. Thermogravimetric analysis and differential scanning calorimetry (TGA-DSC) were performed on a NETZSCH STA 449F3 apparatus. The analyses were performed at a heating rate of 10 ºC/min within the temperature range of 30-400 ºC using a current of nitrogen as inert gas purge (10 mL/min). Solid-state 13C NMR spectra of 1 and 2 were recorded at 298 K on a Varian 300 spectrometer operating at Larmor frequencies of 75.4, 79.3. The 13C experiments were performed with cross-polarization (CP), high power decoupling, and magic angle spinning (MAS) and contact time tCP 1500, using a Bruker double-bearing probe head, spinning speed 4.5 KHz.
2.4 Crystallographic investigations
Powder X ray diffraction experiments were performed in a Bruker D500 diffractometer operated at 40 kV and 35 mA with Cu Kα radiation (1.5406 Å), radius of 217 mm, divergence slit 0.6 mm and anti-scattering slit 0.1 mm, with soller of 2.6º, graphite monocromator and scintillation detector. The range of measurement was from 5-75º.
The relevant details of 2, data collection and structure refinement can be found in Fig. 2. Data collection for 2 was performed at 298 ºC using MoKα radiation on a Bruker APEX II CCD instrument. Semi-empirical absorption correction was applied to all data sets [15]. Structure solutions, refinements, and geometrical calculations were carried out by SHELXTL [16]. The structure was refined using full-matrix least squares methods on F2 with all non-H atoms anisotropically defined. The hydrogen atoms were placed in calculated positions using the “riding model” with Uiso = aUeq (where a is 1.5 for -CH3 and -OH moieties and 1.2 for others).
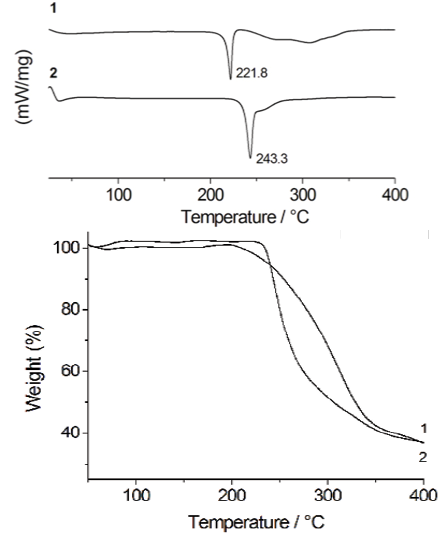
Fig. 1 DSC thermograms of 1 and 2. The peaks in the figure are related to the first thermal event (left). TGA profiles of 1 and 2 (right).
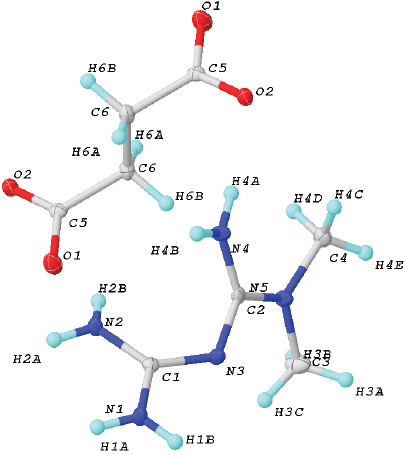
Fig. 2 Unit cell of 2. Ellipsoids were drawn at 50% of probability. Crystal data for (2): formula C6H14N5O2, F.W. 188.22, Monoclinic, P21/n, a 9.5551(3) Å, b 8.8279(3) Å and c 10.8487(3) Å, α 90º, β 92.4900(10)º and γ 90º, V 897.03(5) Å3, Z = 4, ρcalc 1.367 (g cm-3), μ(MoKα) 0.105 mm-1, F(000) 404, crystal size 0.374 Å x 0.22 Å x 0.18 Å mm3, 2Ф range 5.562-50.576º, T 298(2) K, Reflections collected 7485, Independent reflections 1657 [Rint= 0.0282, Rσ= 0.0211], data/restrains/parameters 1657 / 6 / 139, GOF on F2 1.073, Final R indexes [I>= 2σ (I)] R1= 0.0345, wR2= 0.0909, Final R indexes [all data] R1 = 0.0408, wR2 = 0.0971, Largest diff. peak / hole / eÅ-3 0.29/-0.21.
3 Results and Discussion
3.1 DSC-TGA studies
Evidence for the formation of the salt 2 was initially obtained from the DSC and TGA studies. DSC experiments were used to follow the progress of the reaction. The DSC thermograms of 1 and 2 are shown in Fig. 1. The peak of 2 at 243 °C does not correspond to the melting point of either of the starting materials [17-18], indicating the formation of a new solid phase. The relevant thermal data are collected in Table 1.
Table 1 Relevant data from DSC-TG analyses of 1 and 2.
a Peak temperature and ∆Hfus of 1 and 2, is regarding to the first thermal event observed in DSC experiments.
b Crushed single crystals were employed.
Tdecomp of pure disodium succinate, 470 °C.18
Thus the results obtained for 2 clearly show the formation of a new salt with higher thermal stability, properly fitting with those reported for other co-crystals/salts containing carboxylic acid functional moieties [7e]. The DSC profile of 2 exhibits one thermal event at 210-250 ºC due to the melting of the salt. The second process, in the range 250-290 ºC, can be attributed to the decomposition of two H-MET+ cations followed by the decomposition of SUC2-. This fact is supported by simultaneous TGA measurements.
3.2 Crystal structure
The salt 2 crystallizes in the monoclinic P21/c space group (Fig. 2). The asymmetric unit includes one H-MET+ and half molecule of SUC2-, in agreement with the stoichiometry ratio found by DSC-TG analysis (Fig. 2).
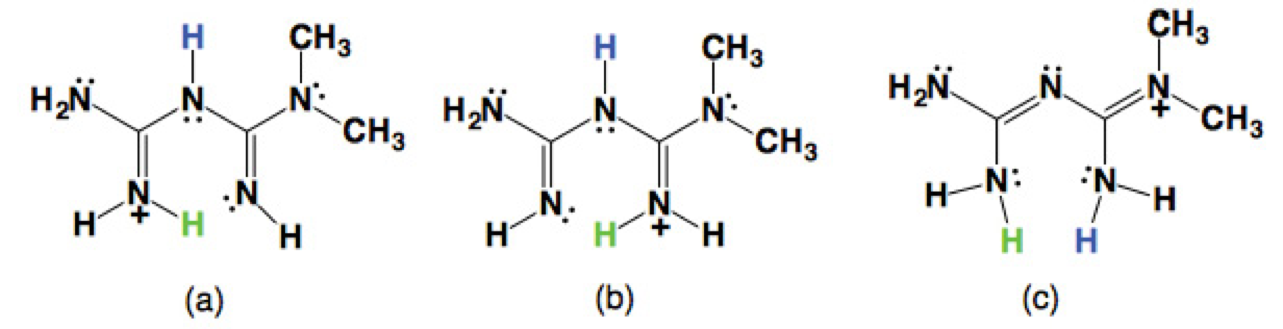
So far, there are three tautomeric forms reported for the cation H-MET+ in the solid state (Scheme 2). In our case, the H-MET+ in 2 corresponds to tautomer (c). Thus, the crystal structure shows the C-N bond distances in H-MET+ to be in the range of 1.330(2)-1.453(2) Å, these values are intermediate between a single and a triple bond [19-20], indicating a delocalized π electron system across the molecule. The H-MET+ cation is non-planar with a torsion angle C3-N2-C4-N5 of 144.55º.
The H-MET+ is linked to SUC2- via different hydrogen bonds with different motifs (Fig. 3). Table 2 contains the geometric parameters for selected hydrogen bonds. The most relevant motifs identified (based on graph set theory [21]) are guanidinium···guanidinium [dimer R2 2(8) motif I], charged-assisted hydrogen bonds guanidinium···carboxylate [dimer R2 2(8) motif II] and N-H···O- (carboxylate) [tetramer R2 4(8) motif III] (Fig. 3, Table 2).

Table 2 Geometric parameters for selected hydrogen bonds of 2.
a Tetramer R2 4(8) and b Dimer R2 2(8).
Motifs I-III produce infinite layers along the a-axis (Fig. 3). In this case, motif I is perfectly aligned between the planes N2-C3-N3-3HA and N2-C3-N3-3HA. However, motif II exhibits a deviation of 25.93º between the plane O2-C1-O1 (carboxylate) and the plane H1B-N1-C3-N3-H3B. A comparison of this value with similar structures deposited at CSD (6.5º aminoguanidinium hydrogensuccinate-GOLXUB [22], 12.45º biguanidium hydrogen succinate-PUNPUK [23], 9.27º guanidinium hydrogen succinate-SEWXAU [24], 6.46º 2-phenylbiguanidinium hydrogen succinate methanol solvate-WADTEC [25]) reveal a significant deviation of motif II in 2. This probably being due to the juxtaposition of motif II with motif III in the crystal lattice (Fig. 3). Thus constraining the spatial approaching of the carboxylate and guanidinium moieties; moreover, in motif III the tetramer R2 4(8) is completely planar.
Other identifiable motifs in the structure, such as N-H···O- (carboxylate) [R2 4(12) formed by N3-H3B···O1 2.051(13) Å, N4-H4A...O1 2.001(12) Å and N4-H4B···O1 2.154(13) Å] and N-H···O- (carboxylate) [R2 2(13) formed by N1-H1A···O2 2.045(15) Å and N4-H4A···O1 2.001(12) Å] allow the extend of the 3D arrangement along the c-axis.
3.3 Spectroscopic analysis
Studying the infrared spectra in two sub-regions allows the identification of hydrogen bonding in the solid state: 3550-3250 cm-1 for N-H stretching vibrations and 1650-1581 cm-1 for N-H deformation vibrations [17, 26]. It has been reported that when hydrogen bonding (X-H···Y) occurs in solid state, the X-H stretching vibrations decrease in frequency and conversely the X-H deformation vibrations shift to higher frequencies [27]. Additionally, hydrogen bonding can be associated with the broadening and enhance of intensity of X-H stretching vibrations [27]. Thus, in order to provide further evidence for the possible presence of motif II, we analyzed comparatively these regions and vibrational bands in the IR spectra of 1 and 2 (Fig. 4, Table 3).

Table 3 Vibrational band assignment for (1-2).
Vibrational modes: a N-H stretching, b asymmetric N-H stretching, c symmetric N-H stretching, d,e N-H deformation. * In parentheses is the absolute value of shifting, | ∆ cm-1 |= vibration mode of 1 - vibration mode of 2.
N-H stretching vibrations, although special attention was placed in the bands at 3368 cm-1 N-H stretching, 3292 cm-1 asymmetric N-H stretching and 3149 cm-1 symmetric N-H stretching of 1 (Table 3) [26], a straightforward comparison with 2 was not possible due to the broadness of the bands found. In the case of the N-H deformation vibrations, for 2 they shift to higher frequencies (Table 4) compared with those of 1 at 1623 and 1555 cm-1 [(26]. This information, in combination with the band at 1650 cm-1 due to large proton polarizability and that due to the unbound -COO- at 1595 cm-1 [(28], may suggest the predominant presence of hydrogen bonding (motif II) in 2 (Table 3).
3.4 Powder X-Ray difraction (PXRD)
The collection of the PXRD measurements of 1-2 is shown in Fig. 5, Na2[SUC] and simulated of 2. The experimental and simulated PXRD measurements of 2 are practically identical, and different when compared to 1 and Na2[SUC] salt.

3.5 ssCPMAS 13C NMR analysis
The ssCPMAS 13C NMR spectra of 1, 2 and Na2[SUC] were determined (Fig. 6, see Scheme 3 for the atom numbering of structure). The spectrum of 1 shows three signals, at δ 38.5, 40.5 and a broad one at δ 157.6 and 160.9 ppm. The up field signals (δ 38.5 and 40.5 ppm) correlate well with the solution NMR data (δ 38.4 ppm in D2O)17. The appearance of the two signals in the solid-state experiment allow to distinguish two different -CH3 groups. This observation agrees well with the results observed in the X-ray crystal structure of Metformin hydrochloride [CSD refcode JAMRIY01, polymorph A20], where one methyl group has the interaction C3-H11···Cl1 [2.897(16) Å and 159.53º] while the other shows short contacts C4-H8···C1 [2.764(17) Å and 143.95º]. The broad downfield signal corresponds to the quaternary carbons: C2 (δ 160.9 ppm) and C3 (δ 157.6 ppm).


For the case of Na2[SUC], assignation of the signals in the CPMAS 13C NMR spectra was straightforward based on comparison with the reported data [29]. The CPMAS 13C spectrum of 2 exhibits five signals, two downfield at δ 34.30, 38.45 and 40.47, which by comparison with the spectra of the precursors, the signal at δ 34.30 ppm corresponds to C4 of succinate anion and the other two intense signals are due to C1. According with the X-ray structure of 2, two distinguishable -CH3 are noted due to short contact C6-H6A···O1 [2.702 Å and 114.10º] and C5-H5B···N1 [2.736 Å and 154.82º]. In addition, at up field two more signals are observed; the broad one, again, composed of two overlapped signals due to C2 and C3. Whilst the signal at δ 181.5 ppm is due to C5 of the succinate anion. No significant ∆δ (-0.07 ppm) is observed in this signal compared with Na2[SUC] (Table 4).
3.6 Dissolution rates profiles
In order to explore the potential of 2, for comparative purposes, we determined the intrinsic dissolution rate constants of 2 and 1 under physiological conditions. The experiments were performed using tablets of 1 and 2 (crushed single crystals were employed), prepared with a hydraulic press at a total force of 220 kg/cm2. The dissolution rates were determined with a Wood’s apparatus with phosphate buffer at pH 6.8. Experiments were carried out by triplicated at 37 ºC under constant stirring (50 rpm) in a constant volume of 900 mL. Determinations were done using a UV-vis spectrometer (Thermo Scientific Evolution 60) monitoring the absorption band at 233 nm using a validated method for MET·HCl [30], permitting quantify the amount of drug dissolved in a giving period of time to establish the dissolution rate profile.
The comparison of intrinsic dissolution profiles of 1 and 2 is shown in Fig. 7. One can easily observe that 1 (Kint = 2.94 mg/min*cm2) shows an initial rapid dissolution and minimal variations of this behavior during the period of time of the determinations, conversely for 2 (Kint = 0.885 mg/min*cm2) the dissolution rate increases constantly, allowing us to conclude that indeed 2 exhibits a sustained-release drug delivery behavior compared with 1. This behavior can be attributed to the markedly difference between the values of hydration energy of chloride anion (ΔG°= −340 kJ mol−1) [30] and succinate (ΔG° = −118 kJ mol−1) [31].

Conclusions
The melting reaction of MET·HCl with dehydrated sodium succinate at 220 °C can be used as a rapid (10 min) and efficient method to produce [MET]2[SUC] 2. The crystal structure of 2 is described for first time, this salt displays a 2:1 stoichiometric ratio where metforminiun H-MET+ and succinate ions are linked via charged-assisted hydrogen bonds of type N-H…O- (carboxylate), this interactions extending into a 2D network. Solid state CPMAS 13C NMR experiments support the formation of the new solid phase of 2. The salt 2 exhibits a sustained-release behavior in water under physiological conditions as compared to the commercial MET·HCl 1. The simplicity of the method reported in this work opens new opportunities for the synthesis of a new series of derivative salts of metfor min. In terms of practical application, this report provides a method that avoids the use of solvents, one of the mayor problems in the pharmaceutical industry, thus potentially greening the production of 2. Although, with some caveats, being in this case the production of NaCl an unwanted by-product particularly when refers to T2DM treatment. However, one can envisage this problem to be easily solved by washing the final product with ethanol [33].

